
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 HyperAI超神经
HyperAI超神经 
作者:彬彬
编辑:李宝珠,三羊
浙江大学与之江实验室研究团队提出了一种基于蛋白质口袋 (protein pocket) 的 3D 分子生成模型——ResGen,与以往最优技术相比,速度提升 8 倍,成功地生成了具有更低结合能和更高多样性的类药物分子。
过去,创新药物的发现往往依赖于古早配方或实验中的偶然事件,例如青霉素。多年来,分子生物学和计算化学的进步,使药物设计模式实现了从盲目筛选到合理设计的转变。
尽管如此,药物研发设计仍然是一个多环节流程,链路长且成本高昂,每一个环节的效率提高都有巨大价值。近年来,随着 AI、大数据等技术的广泛应用,AI 辅助药物设计也在一次次的实验中愈发成熟,AI 正在药物研发的多个环节进行着提效增质的升级改革。
其中,高质量的分子生成模型可以有效提升先导化合物发现的效率。目前,大多分子生成工作都采用了基于配体的方法 (LBMG),然而该方法存在诸多局限性,例如无法考虑分子与靶标之间相互作用模式等。因此研究者们越来越关注基于结构的分子生成 (SBMG,structure-based molecular generative) 的方法,即基于靶标结构进行相应的分子生成。
浙江大学侯廷军教授、谢昌谕教授和之江实验室陈广勇联合团队,提出了一种以蛋白质口袋为条件的 3D 分子生成模型——ResGen。该模型采用并行多尺度建模策略,可以捕捉到蛋白靶点与配体间更高层次的相互作用,并实现更高的计算效率。
分子生成过程被表述为全局自回归和原子自回归,以更好地考虑蛋白质口袋的几何形状。研究结果表明,与现有最先进方法相比,ResGen 生成的分子具有更合理的化学结构,并拥有更好的靶点亲和能力。

获取论文:
https://www.nature.com/articles/s42256-023-00712-7
公众号后台回复「3D 分子生成」获取完整 PDF
数据集:训练集与测试集间的序列相似性小于 40%
该研究使用的训练数据集是 CrossDock2020,该数据集用于蛋白质-小分子相互作用研究,特别是用于评估分子对蛋白质口袋的结合能力。
该数据集的初始数据包含超过 2,200 万个蛋白质-小分子配对 (protein–ligand pairs),为确保训练集与测试集之间的序列相似性小于 40%,研究人员经过筛选,得到了约 10 万个蛋白质-小分子配对,测试集中包含了 100 个蛋白质口袋。
数据集链接:
https://1lh.cc/DjuQrx
ResGen 模型:两个分层自回归
ResGen 模型将以蛋白质口袋感知为条件的分子生成问题,表述为两个尺度的自回归问题,即全局尺度和原子组件尺度。其中,全局自回归 (global autoregression) 是指,ResGen 所生成的每个原子,都是基于之前步骤中生成的分子片段和蛋白质口袋结构;原子自回归 (atomic autoregression) 依次产生新添加的原子坐标和拓扑。
ResGen 可以将完整的分子生成过程分解为分步采样,从而以自回归式方式实现整个分子的生成。此外,为了更好地捕获更高层次的相互作用和降低计算成本,研究团队在这个三维条件生成问题中引入了并行多尺度建模技术。

ResGen 框架示意图
* 图 A 示意:在分子生成的过程中,逐步地确认生长点,添加原子(全局自回归),确认原子的位置,然后添加边(原子自回归)。
* 图 B 示意:口袋和参考分子被表示成原子特征 (vector) 和原子坐标 (scalar)。
* 图 E 示意:分子生成过程。i 中的灰色点云代表新生成的原子,具有位置信息;ii 中的绿色点云,是新生成的原子,补充了原子类型红色圆圈表示每一步的焦点原子( focal atom,生长点),而数字是每个原子成为生长点的概率。
效果验证:优于当前最优模型
一直以来,对于基于蛋白质口袋的 3D 分子生成模型有 2 个广泛应用的检验指标——模型是否学习了配体在不同蛋白质口袋中的特征拓扑分布(即靶点的分子图分布),以及口袋内配体的几何分布(即原子位置和构象的合理性)。
对此,研究团队对 ResGen 和现有的最先进模型进行了系列评估。
对于第一条检验指标,团队评估了针对测试集中的靶点和真实的治疗靶点设计生成的分子的结合能 (binding energies) 和类药性 (drug-like properties)。
对于第二条检验指标,团队设计了构象合理性实验,并分析了蛋白与小分子之间的相互作用模式。
在测试集上生成分子:评估模型泛化能力
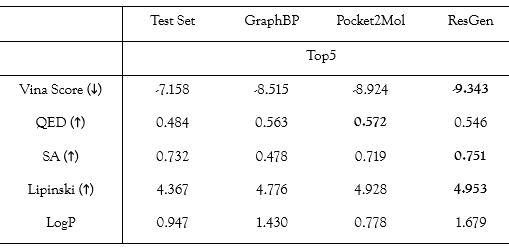
CrossDock 测试集上的 Top5 分子性质
对比结果显示,ResGen 生成的分子在包括结合能和药物相似性在内的大部分指标上都优于 GraphBP 和 Pocket2Mol 生成的分子。
* GraphBP:采用 3D 图神经网络来提取语义信息,然后通过自回归流模型依次生成原子。通过将特定类型和位置的原子逐个放置到给定的结合位点来生成与给定蛋白质结合的 3D 分子。
* Pocket2Mol:用于建模三维蛋白质口袋的化学和几何特征,并采用一种新的高效算法来采样基于口袋条件的新的3D候选药物。
如上图所示,Vina Score 代表了生成分子和对应蛋白靶标的结合能,该指标能够在一定程度上反映模型是否感知到了口袋内的化学环境。
ResGen 在 Vina Score 上的表现意味着,ResGen 更有机会生成和靶标结合更紧密的分子,研究团队认为这可能是因为 ResGen 采用了多尺度建模表征结构,因为这种结构更有利于捕捉蛋白质口袋和配体间更高层次的相互作用(如片段-残基相互作用)。
此外,能否将一个有机化合物推进为候选药物,不仅取决于其与蛋白质相互作用的强度,还取决于它的类药性和可合成性。因此 QED、SA、Lipinski 以及 LogP 这些类药性指标被纳入评估。ResGen 在 SA 和 Lipinski 指标上得分最高,表明 ResGen 更有可能为未认知的蛋白质口袋生成易于合成的类药配体。
针对真实靶标的分子生成:评估现实场景中的表现
为了对模型在真实药物设计场景中的表现进行评估,研究团队以蛋白激酶 B 中 AKT1 和 CDK2 (Cyclin-Dependent Kinase 2) 作为案例,整理了其靶标结构以及具有实验活性的配体化合物,并随机选择了一批无活性小分子作为阴性对照。
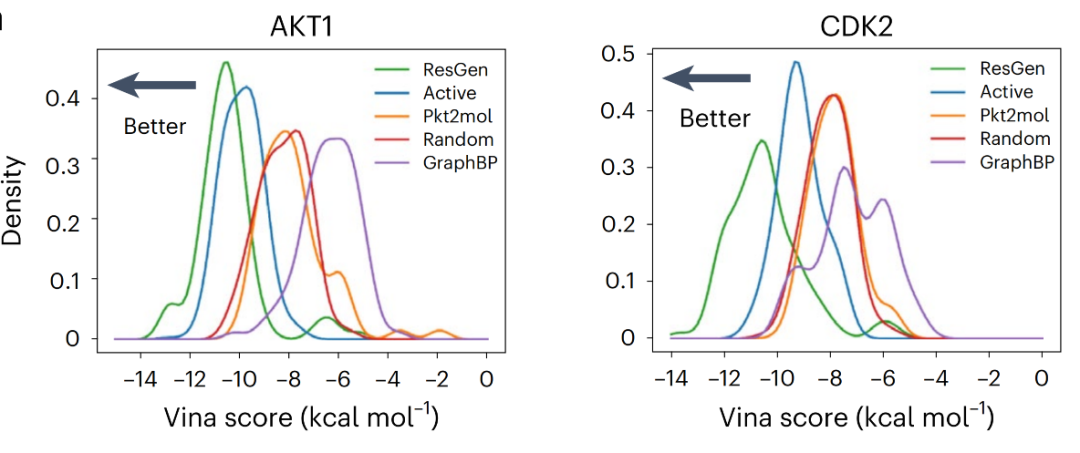
上图展示了各组分子的结合亲和力分布,分布越偏左,结合能绝对值越大,亲和力越高。结果表明,ResGen(绿色) 生成的分子不仅比阴性对照 (Random) 和其他现有最先进模型生成的分子得分更高,而且整体分布甚至略好于 Active。
键长分布实验:评估构象合理性
在构象合理性实验中,研究团队计算了直接生成的分子构象,与传统构像软件生成的分子构象之间的均方根偏差,并比较了生成样本与训练分子之间的键长分布。
在 7 种键长中,ResGen 在 5 种键长中表现最佳,大大优于 GraphBP(大约 10 倍)。与其他两个现有的最先进模型相比,ResGen 能生成更平滑的构象,这突出了其在捕捉蛋白质口袋内部复杂几何分布方面的强大能力。
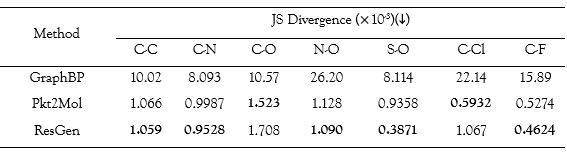
不同方法的键长分布与训练集的键长分布比较
AlphaFold 预测结构分析:评估模型对相互作用的敏感度
为验证 ResGen 是否成功学习了依赖靶点几何结构的相互作用模式,以及模型对蛋白-小分子相互作用的敏感度,研究团队以 X 射线晶体结构和 AlphaFold 预测结构为条件分别生成了两组分子,并对了比这两组分子的结构特征。

基于晶体结构和 AlphaFold 预测结构生成的分子。其中白色配体为共晶配体,X Å 为经过对齐后预测结构与真实结构间的 RMSD。第一列中的白色圆球代表可能的结合位点。
AlphaFold 预测的构象 「封闭 」了晶体构象中存在的口袋,导致模型无法在原口袋位置生成完整的分子,而是在新形成的空腔中生成小片段,表明了 ResGen 的分子生成过程灵敏地依赖于给定的蛋白质口袋。

AlphaFold 预测构象中形成的口袋与晶体口袋相比差异较小,但是模型仍然可以捕捉到这种变化。ResGen 生成的分子更多地占据了 AlphaFold 预测构象中的空腔结构(如图中红圈所示)。
这一实验证明了 ResGen 对靶点结构的敏感性,也暗示了正确的蛋白结构对于 SBMG 策略的重要性。
「AlphaFold2 推理蛋白质结构」详细教程:
https://openbayes.com/console/public/tutorials/m6k2bdSu30C
AlphaFold 蛋白质结构数据集:
https://openbayes.com/console/public/datasets/ETTgyY1oZat/1/overview
点击「阅读原文」即可一键 input,无需下载数据集
侯廷军:致力于计算机辅助药物设计核心问题的研究
分子生成是一项典型的多目标优化任务。我们生成的分子不仅希望他具有好的亲和力,还需要有好的成药性,低的毒性,高的合成性等。
——侯廷军
在传统的药物发现过程中,药物创新存在研发周期长、投入高、风险大等问题。先导化合物的发现和优化是整个药物发现过程中最具挑战性的阶段,需要克服化合物化学空间巨大的难题(可能达到 10 的 60 次方量级);此外,先导物的筛选、优化和评价过程非常复杂。
而通过深度学习和大数据分析,AI 能够高效处理和解读大规模的生物信息学数据,挖掘隐藏在庞大数据集中的模式和关联,提高对潜在药物靶点的识别准确性,加速药物筛选和设计的过程。
面向 AI 辅助药物研发领域,侯廷军教授及其团队长期围绕计算机辅助药物设计中的核心问题,展开前沿交叉学科研究,并取得一系列颇具价值的成果,例如:
* 分子对接虚拟筛选方面,提出新型的基于图表征学习的蛋白-小分子相互作用的打分方法 IGN、基于深度学习的高通量分子对接框架 KarmaDock 等。
*智能分子生成和优化方面,提出基于配体的多约束分子生成方法 MCMG、基于拓扑表面和几何结构的 3D 分子生成方法 SurfGen 等。
*分子成药性及安全性评估方面,提出基于多图注意力模型的毒性预测方法 MGA,成药性预测软件系统 ADMETlab2.0 等。
除此之外,侯廷军教授团队还研发了基于子结构掩盖的 AI 模型可解释性方法 SME,对 AI 模型的可解释性提出了解决方法。
尽管 AI 在药物研发中发挥的巨大价值日益凸显,但作为新兴研究,在实际落地中或许还存在相应的挑战,而这些恰恰也将成为未来的重点研究方向。
对此,侯廷军教授表示,如何有效提升基于 AI 的性质预测方法的预测能力、基于 AI 的打分函数在虚拟筛选中的预测能力、关键成药性参数和毒性终点的预测精度,将是 AI 辅助药物发现领域未来需要重点关注的方向与挑战。
参考资料:https://mp.weixin.qq.com/s/cxpbeGmrHULcWsbVbvQmJA
来源: HyperAI超神经
