
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 HyperAI超神经
HyperAI超神经 
阿尔茨海默病、帕金森病、癫痫…..这些令人「闻名胆颤」的神经退行性疾病是老年健康的隐形杀手,而这类疾病的发生多与蛋白质-RNA 之间的异常结合相关。
在生物医学领域,研究蛋白质-RNA 结合至关重要,因为它在基因表达调控、RNA 加工与剪接、翻译调控以及细胞应激反应等多个生物学过程中发挥着核心作用。理解蛋白质-RNA 结合的机制是揭示复杂基因调控过程和解析疾病的遗传基础的关键,同时,蛋白质-RNA 相互作用在 RNA 靶向治疗中也具有重要应用,为癌症、遗传性疾病及病毒性疾病的治疗提供了新的方向。
近日,在国际人工智能顶会「第 39 届人工智能年会」(The 39th Annual AAAI Conference on Artificial Intelligence, AAAI 2025) 公布的入选成果中,来自清华大学、伦敦大学学院、莫纳什大学、北京邮电大学的联合团队提出的 CoPRA 模型引起了圈内广泛关注,并入选 Oral 环节。
这是首次尝试通过复杂结构架构将蛋白质语言模型 (PLM) 与 RNA 语言模型 (RLM) 结合,用于蛋白质-RNA 结合亲和力预测。为了测试 CoPRA 性能,研究人员从多个数据源整理了最大的蛋白质-RNA 结合亲和力数据集,并在 3 个数据集上评估了模型性能,结果显示 CoPRA 在多个数据集上达到了最先进的性能。
相关成果以「CoPRA: Bridging Cross-domain Pretrained Sequence Models with Complex Structures for Protein-RNA Binding Affinity Prediction」为题,已发布预印本于 arXiv。

论文地址:
https://arxiv.org/abs/2409.03773
CoPRA 仓库地址:
https://github.com/hanrthu/CoPRA
开源项目「awesome-ai4s」汇集了 200 余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
生物医学界持续推进蛋白质-RNA 相互作用研究
过去多年,生物医学界的研究人员一直没有停下过对于蛋白质-RNA 相互作用的研究工作,并且取得了不少进展。
CLIP 实验技术作为 RNA 研究最重要的技术之一,可以解析 RNA 结合蛋白 (RBP) 在整个转录组上的结合图谱,是系统理解一个RBP 功能及其调控机制的基础。但 CLIP 实验费时费力,一次只能提供某一 RBP 在特定细胞环境下的 RNA 结合位点,而且对于实验材料要求较高。然而,蛋白质和 RNA 的结合随着细胞环境的变化可能发生很大的改变,但研究蛋白质对 RNA 的调控需要相同细胞环境的结合信息。
为了解决 RBP 在不同细胞环境下结合动态变化的问题,2021 年 2 月,清华大学结构生物学高精尖创新中心张强锋课题组在 Cell Research 杂志上,发表了题为「Predicting dynamic cellular protein–RNA interactions by deep learning using in vivo RNA structures」的研究成果。该工作使用 icSHAPE 实验解析了 7 种常用细胞类型的 RNA 二级结构图谱,并开发人工智能算法,整合实验获得的细胞内 RNA 结构以及对应细胞环境的 RBP 结合信息,建立了基于细胞内 RNA 结构信息预测细胞内 RBP 动态结合的新方法 PrismNet。
为预测蛋白质-RNA 结合亲和力,业界也有多个计算方法被相继提出,包括基于序列和基于结构的方法。基于序列的方法分别使用不同的序列编码器处理蛋白质和 RNA 序列,并随后建模它们之间的相互作用。然而,由于结合亲和力主要由结合界面的结构决定,这些方法的性能通常受到限制。其他近期提出的方法专注于提取结合界面的结构特征,如能量和接触距离。基于这些提取的特征,研究人员发展出基于结构的机器学习方法,可用于亲和力预测。然而,由于数据集规模的限制,这些方法在新样本上的泛化能力有限,且高度依赖于特征工程。
随着人工智能技术的兴起,不少蛋白质语言模型 (PLMs) 和 RNA 语言模型 (RLMs) 已被开发,这些模型在各种下游任务中展现了出色的性能和泛化能力。同时,由于蛋白质/RNA 的三维结构对于理解其功能至关重要,将结构信息融入语言模型也成为一种新趋势。
比如,美国密苏里大学、肯塔基大学与阿拉巴马大学组成的团队利用多视角对比学习技术将关键的蛋白质结构信息融入到蛋白质语言模型中。基于这个设想,该团队开发了 S-PLM:一种具有蛋白质 3D 结构信息感知能力的蛋白语言模型。S-PLM 在多项蛋白质预测任务中展现出卓越性能,使用轻量化调优工具进行训练后,S-PLM 在蛋白质功能预测、酶反应类别预测和二级结构预测等任务中的性能达到或超过当前最先进的方法。相关研究以「S-PLM: Structure-aware Protein Language Model via Contrastive Learning between Sequence and Structure」为题发表于 bioRxiv。
不过,尽管当前业界的研究展示了结构信息驱动的生物语言模型在交互任务中的巨大潜力,但将来自不同生物学领域的预训练模型结合的工作仍然少见。而在清华大学、伦敦大学学院、莫纳什大学、北京邮电大学联合提出的 CoPRA 中,首次尝试将蛋白质和 RNA 语言模型与复杂结构信息结合,用于蛋白质-RNA 结合亲和力预测。
设计轻量级 Co-Former 模型构建 CoPRA
整体而言, CoPRA 模型的构建过程如下图所示:
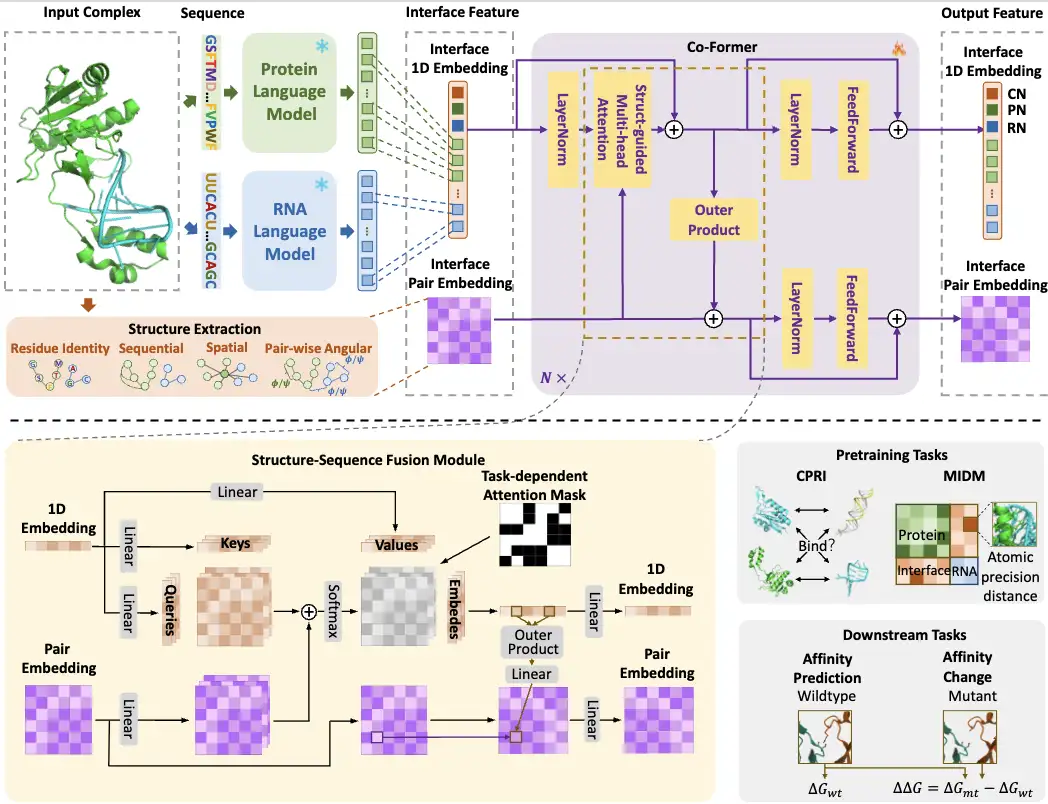
图:CoPRA 模型概述
首先,研究人员将蛋白质和 RNA 序列分别输入到 PLM 和 RLM 中,然后从两个语言模型的输出中选择交互界面处的嵌入,作为后续跨模态学习的序列嵌入。同时,其也从交互界面提取结构信息 (interface feature),作为配对嵌入。
然后,研究人员设计了一个轻量级的 Co-Former 模型,将来自两个语言模型的界面序列嵌入与复杂结构信息结合,形成结构-序列融合模块 (structure-sequence fusion module)。具体而言,Co-Former 通过结构引导的多头自注意力和外积模块融合 1D 和配对嵌入,并应用任务相关的注意力掩码。Co-Former 的输出特殊节点和配对嵌入根据不同任务进行使用,包括两个预训练任务 (Pretraining task) 和两个下游亲和力任务 (Downstream task)。
研究人员还为 Co-Former 提出了一个双范围预训练策略,以建模粗粒度的对比交互分类 (CPRI) 和精细粒度的界面距离预测 (MIDM),以原子级精度进行学习。
为了评估 CoPRA 与其他模型的性能,研究人员需要解决统一标注标准数据集缺失的问题。于是,他们从 3 个公共数据集收集了样本:PDBbind、PRBABv2 和 ProNAB,整理了最大的蛋白质-RNA 结合亲和力数据集 PRA310,并在 PRA310 和 PRA201 数据集上评估了其模型性预测蛋白质-RNA 结合亲和力的能力。
*PRA201 数据集:PRA310 的子集,每个复合物仅包含一个蛋白链和一个 RNA 链,并且有更严格的长度限制
CoPRA 在预测蛋白质-RNA 结合亲和力方面性能最佳
如下表所示,CoPRA 的从头训练版本在 PRA310 数据集上达到了最佳性能。此外,大多数使用 LM 嵌入作为输入的方法表现优于其他方法,表明结合预训练单模态 LMs 进行亲和力预测的巨大潜力。
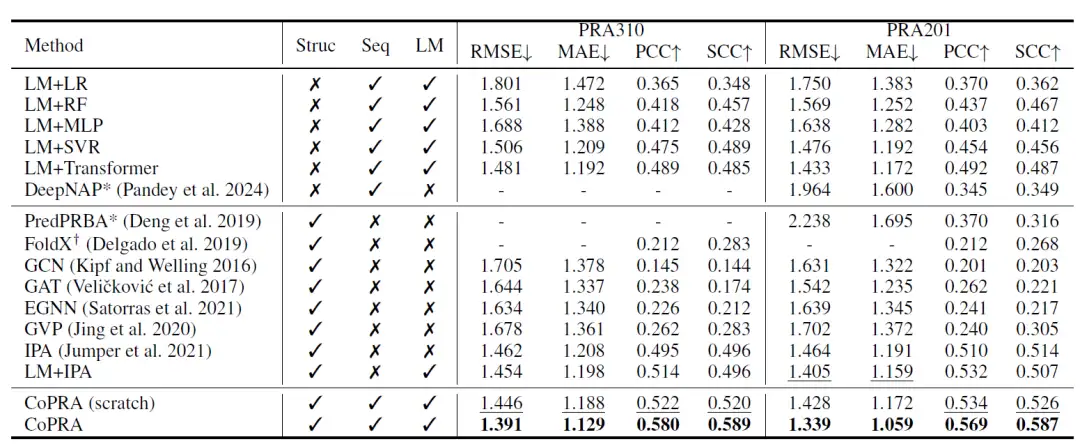
表:PRA310 和 PRA201 数据集上的 5 折交叉验证的平均指标
随后,研究人员使用其整理的无监督数据集 PRI30k 对模型进行了预训练,显著提高了其在两个数据集上的整体性能。在 PRA310 数据集上,CoPRA 的 RMSE 为 1.391,MAE 为 1.129,PCC 为 0.580,SCC 为 0.589,远优于第二好的模型 CoPRA (从头训练版本)。PredPRBA 和 DeepNAP 支持蛋白质-RNA 对亲和力预测,研究人员将这些方法在 PRA201 数据集上的表现进行了比较,结果显示,尽管 PRA201 中至少有 100 个样本出现在它们的训练集中,但它们在 PRA201 上的性能明显低于它们报告的结果,表明这些方法的泛化能力较差。
CoPRA 在预测突变对结合亲和力影响方面更强,且泛化能力极佳
为了进一步评估模型对亲和力的细粒度理解,研究人员将模型重定向为预测蛋白质的单点突变对蛋白质-RNA 复合物的影响。参考蛋白质突变效应预测的相关研究,研究人员在每个复合物级别上对指标进行平均,评估了 CoPRA 在 PRI30k 上进行预训练并在 PRA310 上进行调优后的 zero-shot 性能和微调性能。
如下表所示,在使用 mCSM 的交叉验证集进行微调后,本研究提出的模型在所有 4 个指标上均超越了其他模型,RMSE 为 0.957,MAE 为 0.833,PCC 为 0.550,SCC 为 0.570。
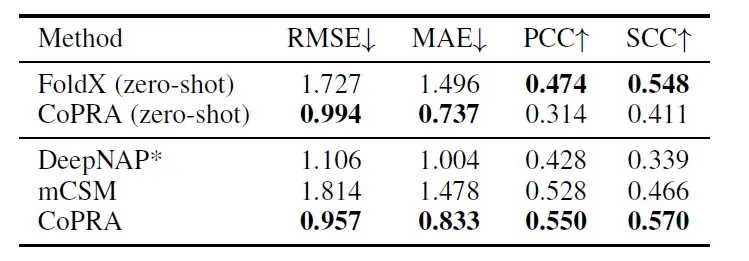
表:在 mCSM 盲测集上的每个结构的表现
尽管没有看到任何突变复合物结构,但这种优越的性能来源于双重预训练目标,这一表现证明了 CoPRA 在不同亲和力相关任务上的泛化能力。
多模态蛋白质语言模型的突破性进展
上文介绍的研究思路本质是将蛋白质、RNA 等多种生物模态与复杂结构信息相结合,也就是所谓的多模态学习 (MultiModal Learning)。简单来讲,多模态学习是在深度学习的框架下,将各种不同类型的数据整合在一个模型中进行建模。
过去几年,随着大语言模型的快速发展,研究人员开始尝试将其应用于蛋白质科学领域,以准确理解和预测蛋白质的功能、结构和性质。然而,此前的蛋白质导向型大语言模型主要将氨基酸序列作为文本形式处理,未能充分利用蛋白质的丰富结构信息,如今,多模态学习的进展则为越来越多的相关研究提供了新思路。
比如,在药物研发领域,准确有效地预测蛋白质与配体的结合亲和力对于药物筛选和优化至关重要。然而,此前的研究没有考虑到分子表面信息在蛋白质-配体相互作用中的重要作用。基于此,来自厦门大学的研究人员提出了一种新颖的多模态特征提取 (MFE) 框架,该框架首次结合了蛋白质表面、3D 结构和序列的信息,并使用交叉注意机制进行不同模态之间的特征对齐。实验结果表明,该方法在预测蛋白质-配体结合亲和力方面取得了最先进的性能,相关研究以「Surface-based multimodal protein–ligand binding affinity prediction」为题,于 2024 年 6 月发布在 Bioinformatics 上。
2024 年 12 月,来自华东师范大学等机构的研究团队提出了一个创新性的解决方案 EvoLLama,这是一个将蛋白质结构编码器、序列编码器和大语言模型进行多模态融合的框架。在零样本设置下,EvoLLama 展现出了强大的泛化能力,相比其他微调基线模型提升 1%-8% 的性能,超越当前最先进的监督微调模型平均 6% 的性能。相关研究成果以「EvoLlama: Enhancing LLMs’ Understanding of Proteins via Multimodal Structure and Sequence Representations」为题已发布预印本于 arXiv。
当然,多模态学习只是可供选择的研究思路之一,未来,通过更多机器学习手段研究蛋白质的表面,生物学家可以更深入地了解其如何与其他生物分子相互作用,从而为新药研发提供助益。
来源: HyperAI超神经
