
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 大学生快乐科普驿站
大学生快乐科普驿站 在古老的地中海沿岸,有一个关于地中海贫血的传说。那里的人们相信,血液里流淌着海神的祝福,而红色的血液是太阳与海洋交汇的精华。然而,一场意外让部分孩子的血脉变得脆弱,无法承载充足的生命能量,他们变得虚弱、苍白。不能像其他人一样在海边奔跑、潜水、捕捉鱼虾,而是常常倚靠在母亲的怀抱里。这个美丽而忧伤的故事,实际上是对地中海贫血的形象化描述。今天我们就来揭开地中海贫血的神秘面纱。
地中海贫血:简称地贫,简单来说,我们的血液里有个重要的“运输工”——血红蛋白,它负责把氧气送到全身。血红蛋白由“珠蛋白链”组成,好比一辆车的零件。如果零件有缺陷,车就跑不动。同样地,珠蛋白链出问题,就会导致红细胞寿命缩短、携氧能力差,不能很好地运输,最终引发贫血。
地中海贫血发病率较高,是我国罕见病中最常见的类型。它不传染但会遗传。在我国大约有 3000万人携带地贫基因,中间型、重型患者大约 30万人,其中重型β地贫患者1.5万人。
一、地中海贫血有哪些类型?
主要分为两大类:
α型:问题出在α珠蛋白链。严重时胎儿往往难以存活,叫作“血红蛋白Bart’s水肿胎综合征”。
β型:问题出在β珠蛋白链。重型β地贫是我们常说的“重型地中海贫血”,多在宝宝出生几个月后(3~12月龄)发病。
简单理解就是α出事,胎儿难活;β出事,婴儿遭殃。
二、这种病是怎么遗传的?我家孩子会不会得?
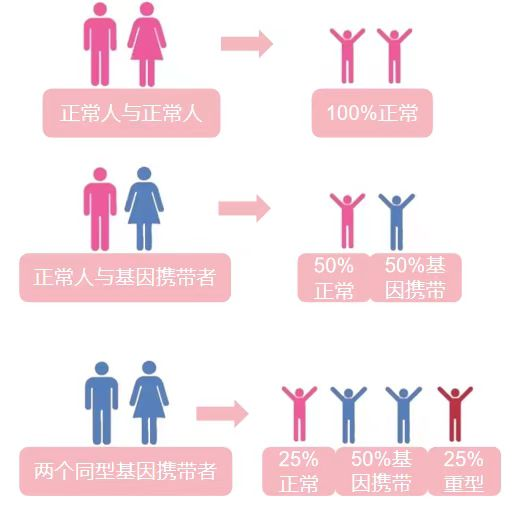
这是一个常染色体隐性遗传病,是否发病取决于父母是否携带缺陷基因
1.假如父母只有一方为携带者:那么孩子不会患重型地贫,有50%可能成为携带者,50%正常。
2.假如父母都是“携带者”,那么孩子有25%的概率是重型患者;50%的概率是携带者(本身没事,但会传下去);还有25%的概率完全正常。
这就像玩“遗传的抽奖游戏”,可惜中奖的不是奖品,而是疾病。
三、为什么会生病?
核心原因:造血不平衡。
1.α或β链造得少 → 另一条链没搭档 → 在红细胞里“胡乱堆积” → 红细胞被损坏。结果就是:红细胞还没上岗,就提前“下岗”了。于是出现两大问题:
溶血(红细胞寿命太短,血红蛋白掉下去)。
无效造血(骨髓再努力生产,但生产线总出次品)。久而久之,孩子会表现出严重贫血、骨骼变形、脾大,还可能造成机体铁过载,引起脏器功能损伤、生长发育迟缓,严重者可能危及生命。
四、重型地中海贫血孩子有哪些症状?
宝宝期(3~12个月):原本健康,突然脸色苍白、乏力、发育落后。逐渐:肝脾肿大,常感染,免疫力下降。外貌改变:额头高耸、颧骨突出、鼻梁塌陷,两眼距离变宽,称为“地贫面容”。长期后果:心脏、肝脏因铁过载受损,可能出现糖尿病、骨质疏松、生长发育迟缓等。如果不治疗,重型β地贫的孩子多在5岁前夭折。

五、怎么检查才能确诊?
1.血常规:通过检测红细胞数量、血红蛋白水平和红细胞形态,初步判断是否贫血。
2.血红蛋白电泳:重型β地中海贫血首诊时血红蛋白电泳显示胎儿血红蛋白显著增高。
3.基因检测:最精准的方法,能直接找到哪条基因出错。
4.产前诊断:如果夫妻双方均为携带者,可在孕期通过绒毛取样或羊水穿刺检测胎儿是否患病。
六、地中海贫血和缺铁性贫血一样吗?
这是个常见误区。虽然两者都叫“贫血”,但有明显区别:
缺铁性贫血:因为缺铁,补铁后就能改善。
地中海贫血:基因问题,补铁没用,反而会“火上浇油”,加重铁过载。所以,别随便给孩子乱补铁,尤其在南方高发区。
七、目前的治疗办法有哪些?
目前针对地中海贫血(重型)有三大“武器”:
1. 规律输血 + 祛铁治疗
输血:定期输血,维持血红蛋白正常,让孩子能正常成长。
祛铁:输血次数多了,铁会在体内堆积,必须用药物把多余的铁排出去。这是目前最常用的方法,但需要终生维持。
2. 造血干细胞移植
是目前治愈地中海贫血(重型)的主要方法。原理:用健康供者的造血干细胞替换患者的“问题骨髓”。
如果成功,可以算是“治愈”。局限性:要找到配型合适的供者,而且费用高、风险大。
3. 基因治疗(正在路上 )
思路:把“坏掉的基因”修好,或给血红蛋白链“加个替补”。
目前仍在临床试验阶段,未来有望成为“终极解决方案”。

八、重点来了!地贫可以防!
虽然地中海贫血难治,但我们可以靠“三级预防”去阻断它。
一级预防:婚前和孕前筛查,父母双方筛查血常规,发现血红蛋白偏低时,建议行基因检测,明确夫妻双方是否携带同型基因。
二级预防:产前诊断,如果夫妻双方都是携带者,可在孕早期进行基因检测,若胎儿确诊重型地中海贫血,建议及时终止妊娠。免孩子出生后痛苦。这就是“把病堵在摇篮里”。
三级预防:新生儿筛查,早诊断、早治疗,改善患儿生存质量。

九、患者需要什么样的长期管理?
1.定期复查血红蛋白和铁指标;
2.规范输血,避免不安全的血源;
3.阶段监测生长发育及内分泌系统水平
4.心理关怀:长期治疗对孩子和家庭都是巨大的考验,需要医护、心理师和社工的支持。
总结:地中海贫血虽然是“遗传密码”里的“失误”,但我们可以用科学的钥匙,去提前解锁它。早筛查、早发现、早干预,就能为下一代筑起一道健康防线,不让悲剧重演,让孩子健康成长!

参考文献:
[1]中华医学会儿科学分会血液学组,《中华儿科杂志》编辑委员会. 重型β地中海贫血的 诊断和治疗指南(2017年版).中华儿科杂志,2018,56 (10):724-729.
[2]中华医学会血液学分会红细胞疾病(贫血)学组. 中国输血依赖型β地中海贫血诊断与 治疗指南(2022年版).中华血液学杂志,2022,43(11):889-896.
[3]中华医学会医学遗传学分会遗传病临床实践指南撰写组,商璇,吴学东,等.β-地中海贫血 的临床实践指南.中华医学遗传学杂志,2020,37(3):243-251. [4]中华医学会医学遗传学分会遗传病临床实践指南撰写组,商璇,张新华,等.α-地中海贫血的临床实践指南.中华医学遗传学杂志,2020,37(3):235-242.
[5]程涛.中国重型β地中海贫血患者疾病负担及诊疗现况横断面调查研究报告. 北京:中华医学电子音像出版社,2023.
[6]蒋海山,彭莹.地中海贫血携带者筛查及防控专家共识[J].国际神经病学神经外科学杂志,2024,51(05):43-50.DOI:10.16636/j.cnki.jinn.1673-2642.2024.05.006.
[7]WANG WD, HU F, ZHOU DH, et al. Thalassaemia in China. Blood Rev, 2023,60:101074.
[8]PEPE A, PISTOIAL, GAMBERINIMR, et al. Nationalnetworking in rare diseases and reduction of cardiac burden in thalassemia major. Eur Heart J, 2022,43(26):2482-2492.
[9]CAPPELLINI MD, VIPRAKASIT V, TAHER AT,et al. A phase 3 trial of luspatercept in 470 patients with transfusion-dependent β-thalassemia. N Engl J Med, 2020,382(13):1219-1231.
[10]THOMPSON AA, WALTERS MC, KWIATKOWSKI J, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med, 2018,378(16):1479-1493.
[11] FRANGOULH, ALTSHULER D, CAPPELLINIMD,et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med, 2021,384(3):252-260.
戴红燕 主管护师 武汉儿童医院
中国医药教育协会科普健康教育工作会委员
亚太儿科叙事医学与健康人文会委员
中国民族卫生协会卫生健康技术推广专家会委员
湖北省血液净化专科护士 武汉儿童医院重症医学科专科护士
第一作者发表论文5篇,实用新型专利专利3项,参编书籍两部,撰写科普文章二十余篇

来源: 武汉儿童医院
