
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
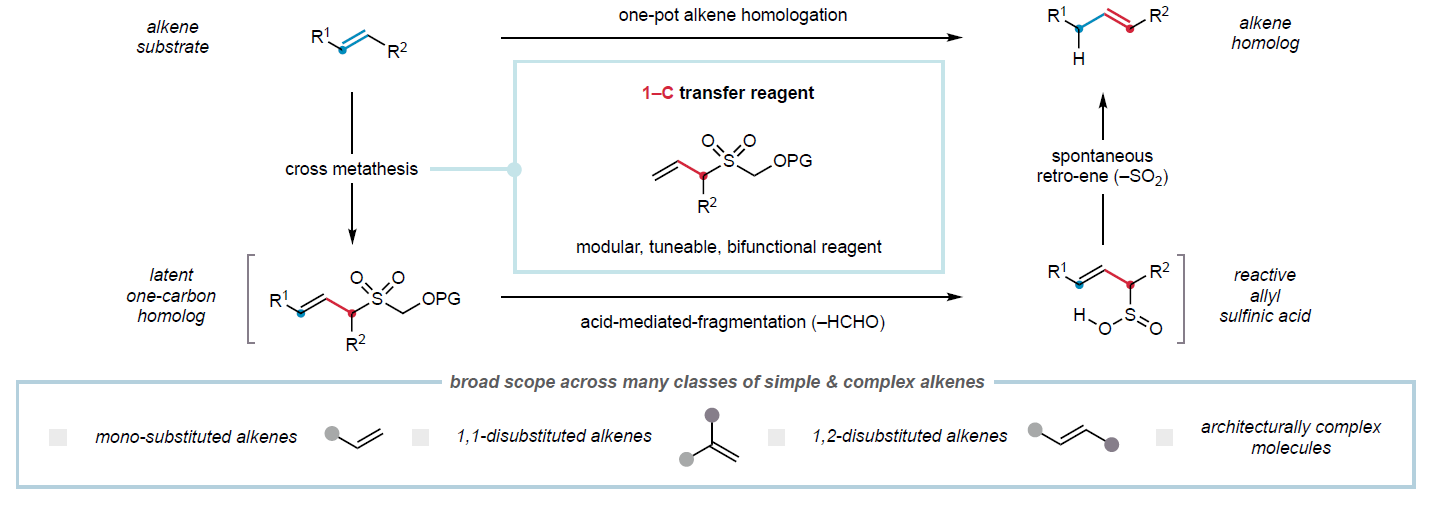
近日,英国剑桥大学(University of Cambridge)Matthew J. Gaunt课题组报道了一种适用于各类烯烃的催化一碳同系化方法。其通过使用新型多功能烯丙基砜试剂的固有反应性,利用一锅法经由串联的交叉复分解和断裂/逆ene反应,实现了烯烃中亚甲基单元的形式插入。此外,作者将该策略应用于多个结构复杂分子的一碳同系化反应中,包括通过这一实用转化获得cyclosporine-A同系物。这些同系物表现出可调控的药理与生物特性,有望成为亲环蛋白抑制剂的新型先导化合物。文章链接DOI:10.1038/s41586-025-09159-9。
(图片来源:Nature)
正文
一碳同系物是指碳链长度相差一个亚甲基单元、结构相关且功能相似的有机分子。在药物分子、天然产物、农用化学品、香料及石油产品等众多类化合物中,同系物间微妙的理化性质差异可导致其功能显著不同。因此,高效合成分子的同系物已成为分子发现计划的重要策略。尽管目前已发展的方法可实现部分官能团的同系化,但烯烃的直接普适性碳链延伸策略仍未有报道。最近,英国剑桥大学Matthew J. Gaunt课题组报道了一种普适的烯烃一碳同系化方法。其利用烯丙基砜试剂,通过交叉复分解和断裂/逆烯反应级联实现了烯烃中亚甲基单元的形式插入。此外,该技术可成功实现cyclosporine-A的同系化,具有良好的实用价值(Figure 1)。 欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
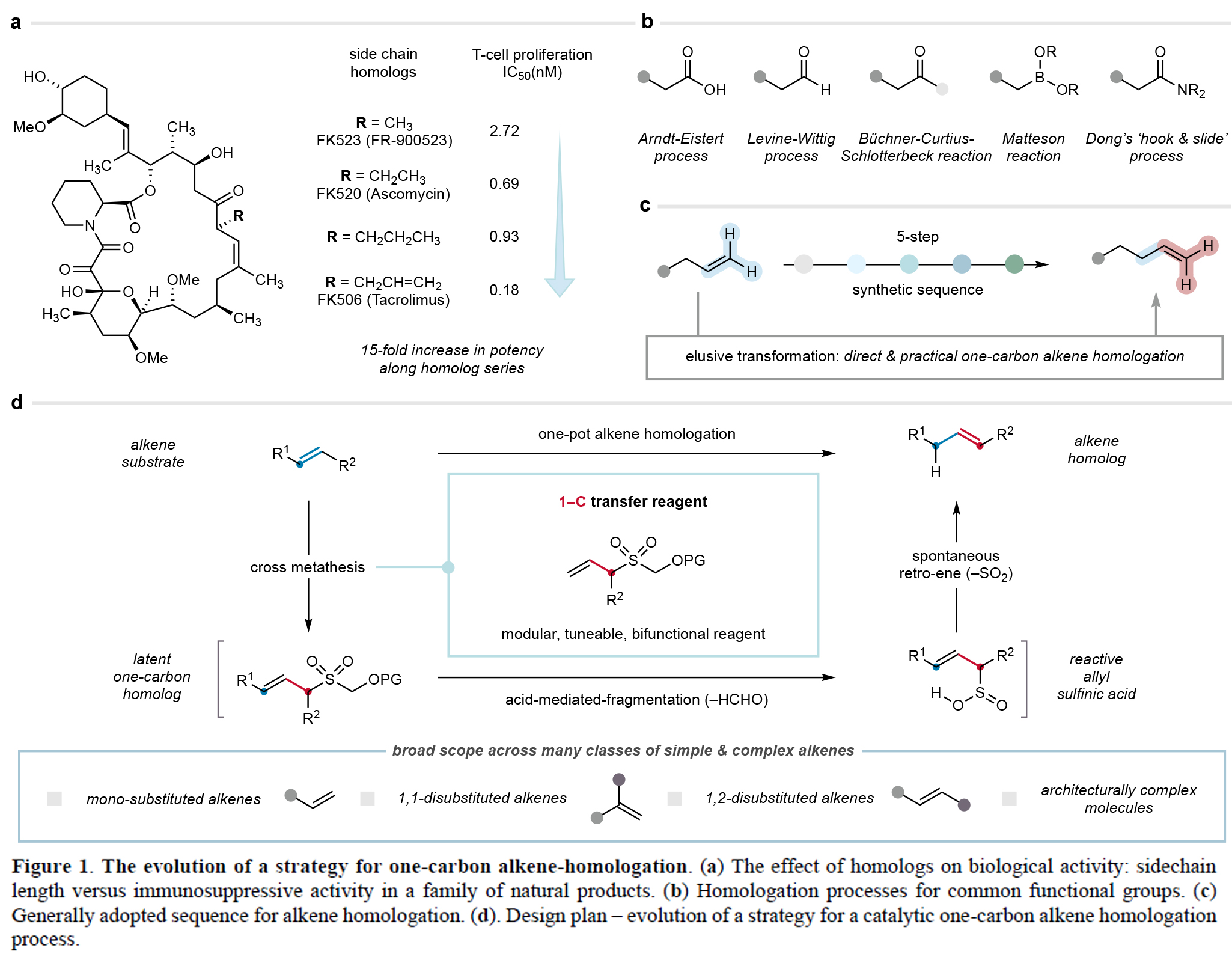
(图片来源:Nature)
首先,作者以烯丙基砜试剂1a和烯烃2a作为模板底物对反应条件进行了考察(Figure 2a)。通过一系列条件筛选,作者发现当使用1a (4.0 equiv), 2a (1.0 equiv), Hoveyda-Grubbs二代催化剂(HG-II)(5 mol%),在DCM中,40 oC反应8小时可以得到3a。随后在2 M MeOH-HCl溶液中搅拌16小时可以以接近定量的产率得到同系化的烯烃产物4a。
在得到了最优反应条件后,作者对此转化的兼容性进行了考察(Figure 2b)。实验结果表明此转化对大多数烯烃底物有效,但对含酯基(4k)和氨基甲酸酯(4o)等酸敏感基团的烯烃,则需采用四丁基氟化铵(TBAF)和柠檬酸的温和逆ene反应条件。其中比较典型的例子包括烯烃4p的合成,该分子若通过传统方法需多步合成。苯乙烯类底物在改良的逆ene条件(90 oC, TFA-H2O)下可高效发生去共轭反应,生成烯丙基苯衍生物4q-4s。含氮杂环(4t)和羧酸(4u)等具有极性官能团的烯烃也能以良好收率完成一锅法同系化,证实了该方法优异的官能团兼容性。值得注意的是,该策略成功应用于多种天然产物,包括sclareol和forskolin以及allylestrenol等均可实现同系化,以26-75%的产率得到相应的产物5-7,且这些生物活性分子的同系物均属首次报道(Figure 2c)。
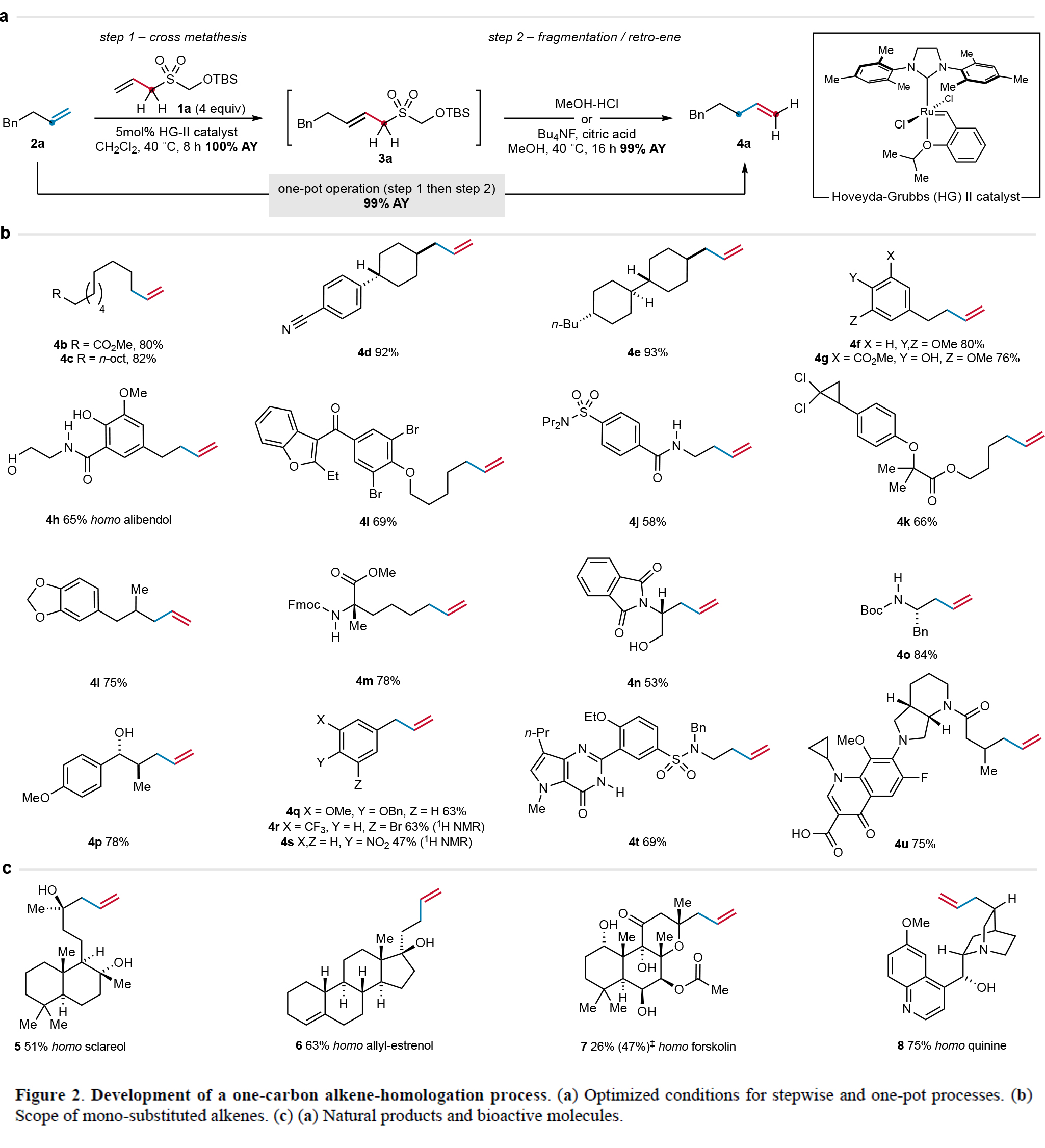
(图片来源:Nature)
本研究成功将一碳同系化策略应用于复杂药物分子改造,在格拉瑞韦(Grazoprevir)中实现ACCA(1-amino-2-vinyl cyclopropane-carboxamide)药效团向烯丙基环丙烷类似物9(43%)的高效转化,并一步完成了FK506同系物10合成(53%),较传统7步路线显著简化。该策略不仅解决了特殊结构的合成难题,更为药物结构修饰提供了高效新途径(Figure 3)。
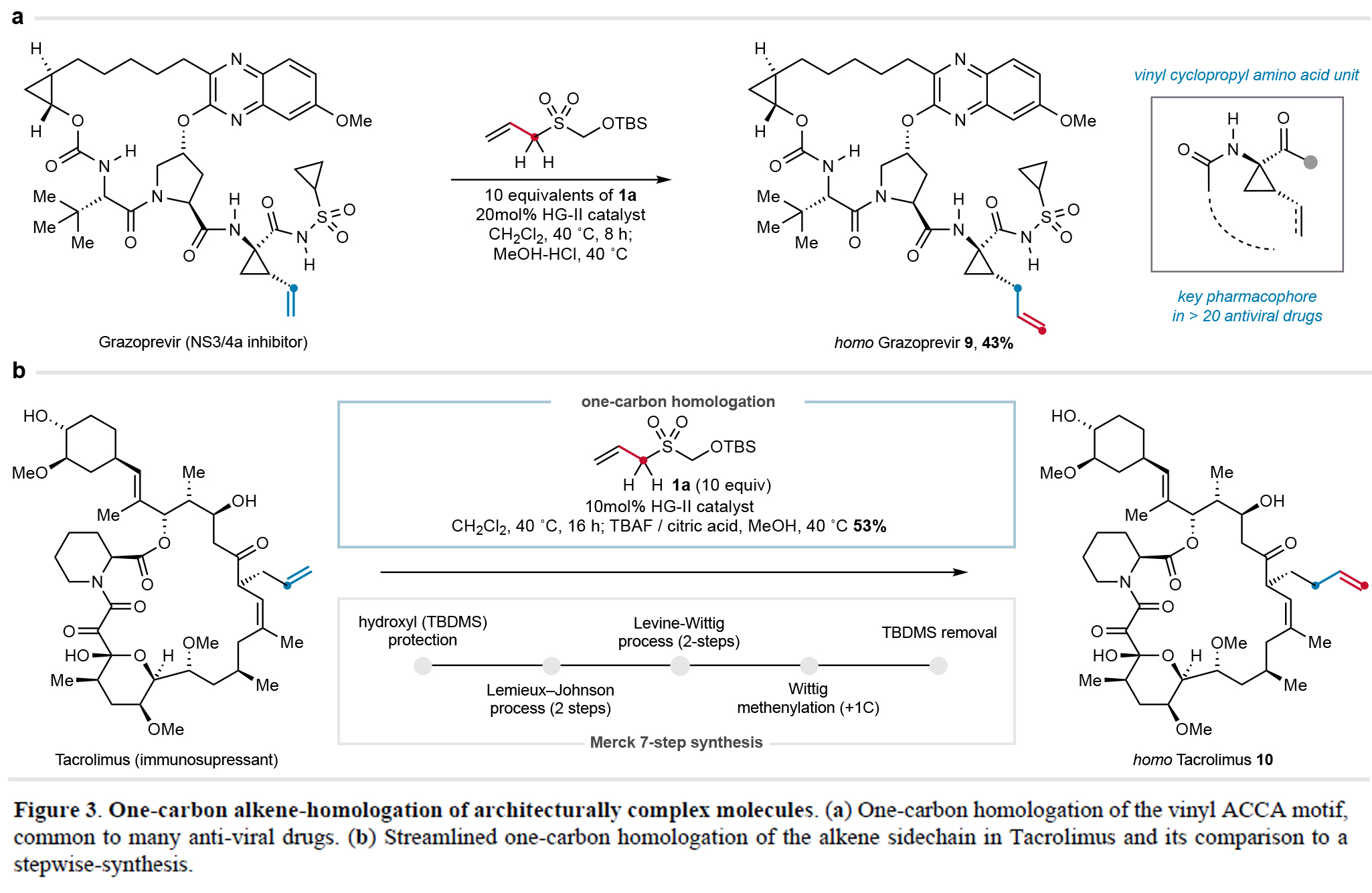
(图片来源:Nature)
本研究成功将一碳同系化策略拓展至二取代烯烃的修饰。通过优化1CTR的结构(如引入偕二甲基),可以实现鱼藤酮等1,1-二取代烯烃的高效同系化(收率最高94%),且获得了具有农用化学品和香料应用价值的α-支链产物(11a-11d)。特别值得关注的是,该方法首次实现了1,2-二取代烯烃的直接同系化。利用甲基修饰的1CTR-1c与氨基酸衍生的烯烃反应,可以以87%收率(E/Z=3:1)得到内烯烃同系物14a。应用异丙基变体1CTR-1d对天然辣椒素提取物进行同系化,成功将同型辣椒素含量提升至45%(15,E/Z = 5:1),该产物较辣椒素具有显著不同的辛辣度,在肿瘤学和镇痛领域具有应用潜力(Figure 4)。
进一步研究表明,该策略还可实现大环烯烃的扩环。通过1CTR-1a介导的开环交叉复分解/RCM,作者可以成功将17元环内酯Ambrettolide高效转化为19元大环20(74%),且可通过催化剂选择调控E/Z构型。机理研究发现,逆ene反应中烯烃几何构型不完全和前体一致,这可能与过渡态中1,3-双直立键相互作用有关(Figure 4)。
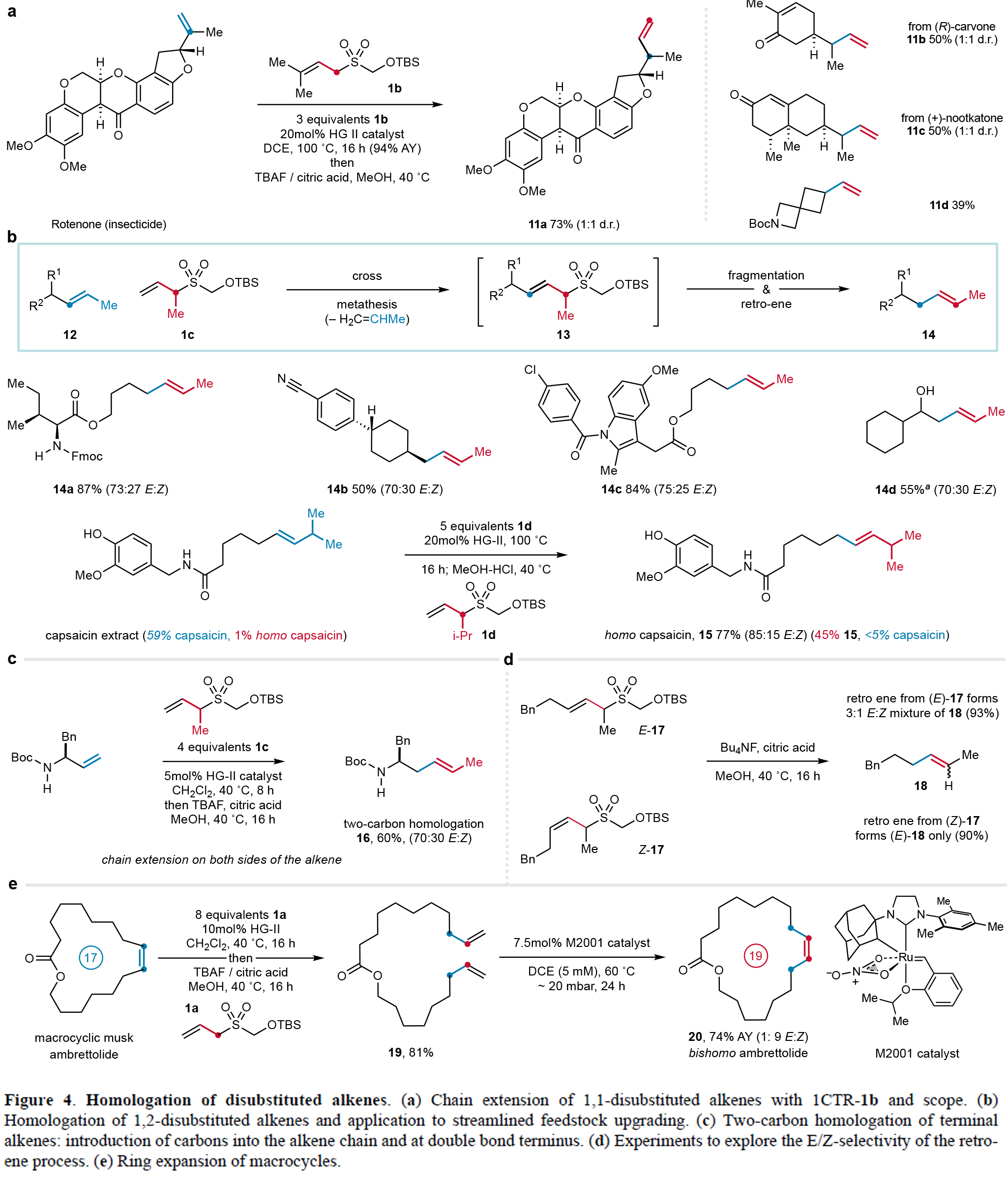
(图片来源:Nature)
此外,此一碳同系化策略可成功应用于cyclosporine-A的结构改造(Figure 5)。通过优化1CTR-1c的反应条件,作者实现了该免疫抑制大环肽烯烃侧链的一碳(21)和两碳延伸(22),产物均以良好收率及E-构型选择性获得(Figure 5a)。生物学评价显示,在 ≤ 320 nM浓度下,一碳同系物21的免疫抑制活性较母体降低13倍,其与cyclophilin-A形成的二元复合物对钙调磷酸酶(calcineurin)的抑制活性也减弱3.5倍(Figure 5b,5c)。而两碳同系物22则保持了与cyclophilin-A相当的活性。这些发现表明,通过精确调控烯烃侧链长度可选择性改变三元复合物的相互作用强度,为开发具有差异免疫抑制活性的cyclosporine类似物提供了新的策略。
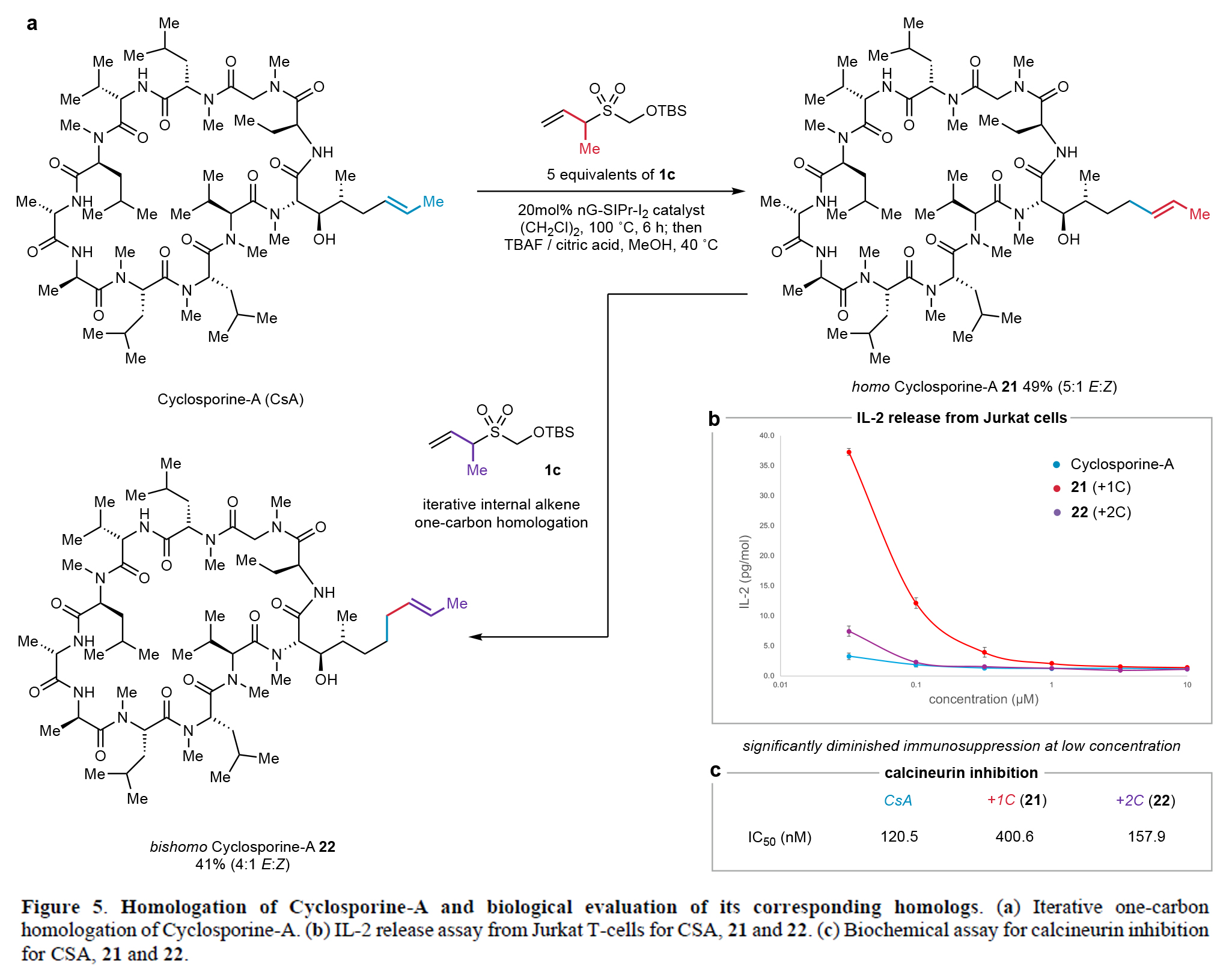
(图片来源:Nature)
基于cyclosporine-A同系物21所表现出的独特免疫抑制活性变化,作者进一步探索了烯烃侧链的精细修饰策略(Figure 6)。利用1CTR的模块化特性,作者成功合成了含氘代甲基(23,E/Z = 5:1)和氟原子(24,E/Z = 1:4)的cyclosporine类似物,其中23实现了甲基到CD3的替换,而24则保持了碳数不变但引入了末端氟取代(Figure 6a)。特别值得关注的是,使用未取代的1CTR-1a通过异构化策略可以以良好收率获得末端烯烃类似物异环孢素A(25),并可进一步同系化为高异环孢素A(26)(Figure 6b)。此外,钙调磷酸酶抑制实验表明,氟化类似物24的活性较母体降低10倍,末端烯烃25降低4倍,而26则恢复至cyclosporine-A的水平(Figure 6c)。TR-FRET实验证实所有类似物(22-26)均保持对亲环蛋白A的高亲和力(<80 nM)。
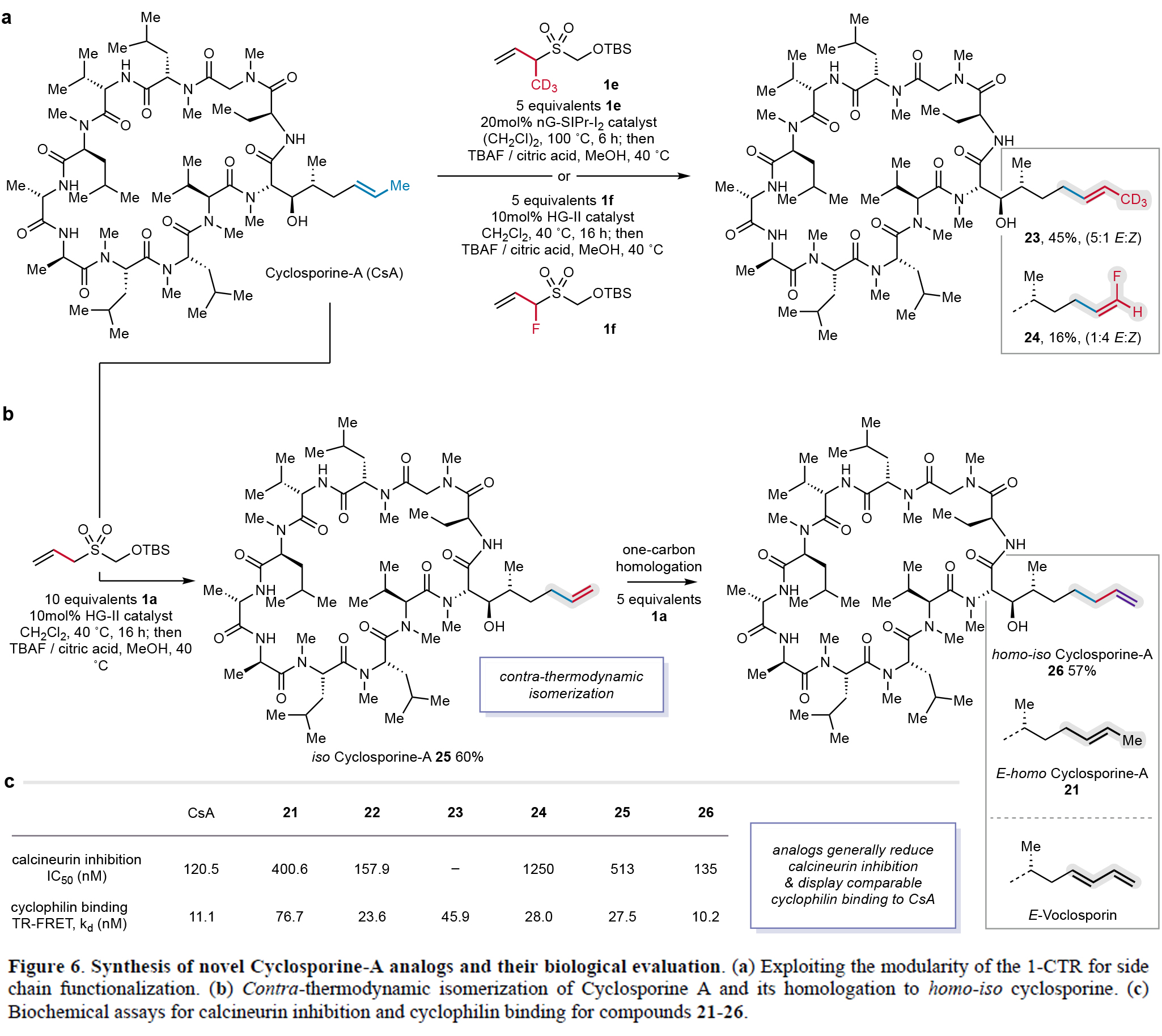
(图片来源:Nature)
总结
传统合成方法的局限性使得复杂分子中精细的系统性结构修饰难以实现,从而导致许多潜在的结构-活性关系和生物学突破被长期忽视。Matthew J. Gaunt课题组开发的烯烃同系化策略为此提供了高效解决方案。其通过可编程的烯烃侧链长度调控,成功获得了一系列结构精确调控的Cyclosporine-A衍生物。该策略可拓展至其他钙调磷酸酶抑制剂,其催化烯烃同系化方法与互补性异构化方法的结合,为构建特定结构的复杂分子提供了全新的工具,在学术研究与工业应用中均具有广阔前景。
文献详情:
One-carbon homologation of alkenes.
Marcus C. Grocott, Matthew J. Gaunt*.
Nature,2025
DOI:10.1038/s41586-025-09159-9
来源: 化学加
