
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
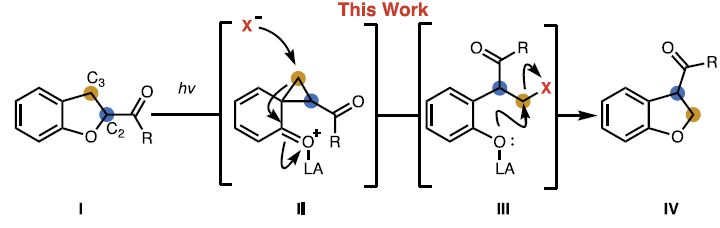
近日,美国加州大学伯克利分校(University of California, Berkeley)Richmond Sarpong课题组报道了通过C2-酰化二氢苯并呋喃的C2-C3位光化学交换反应,实现了形式上的1,2-酰基转位。该策略基于二氢苯并呋喃骨架发生非常规的光化学异构化,生成高亲电性的螺环丙烷中间体,随后被卤素亲核试剂捕获。实验表明,当使用370纳米光照时可实现多种芳基酮的转位;而使用310纳米的光照时则能实现羧酸、酯类和酰胺类化合物的转位。此工作展示了骨架重排策略在实现分子外围修饰方面的强大潜力。文章链接DOI:10.1126/science.adv9915。
(图片来源:Science)
正文
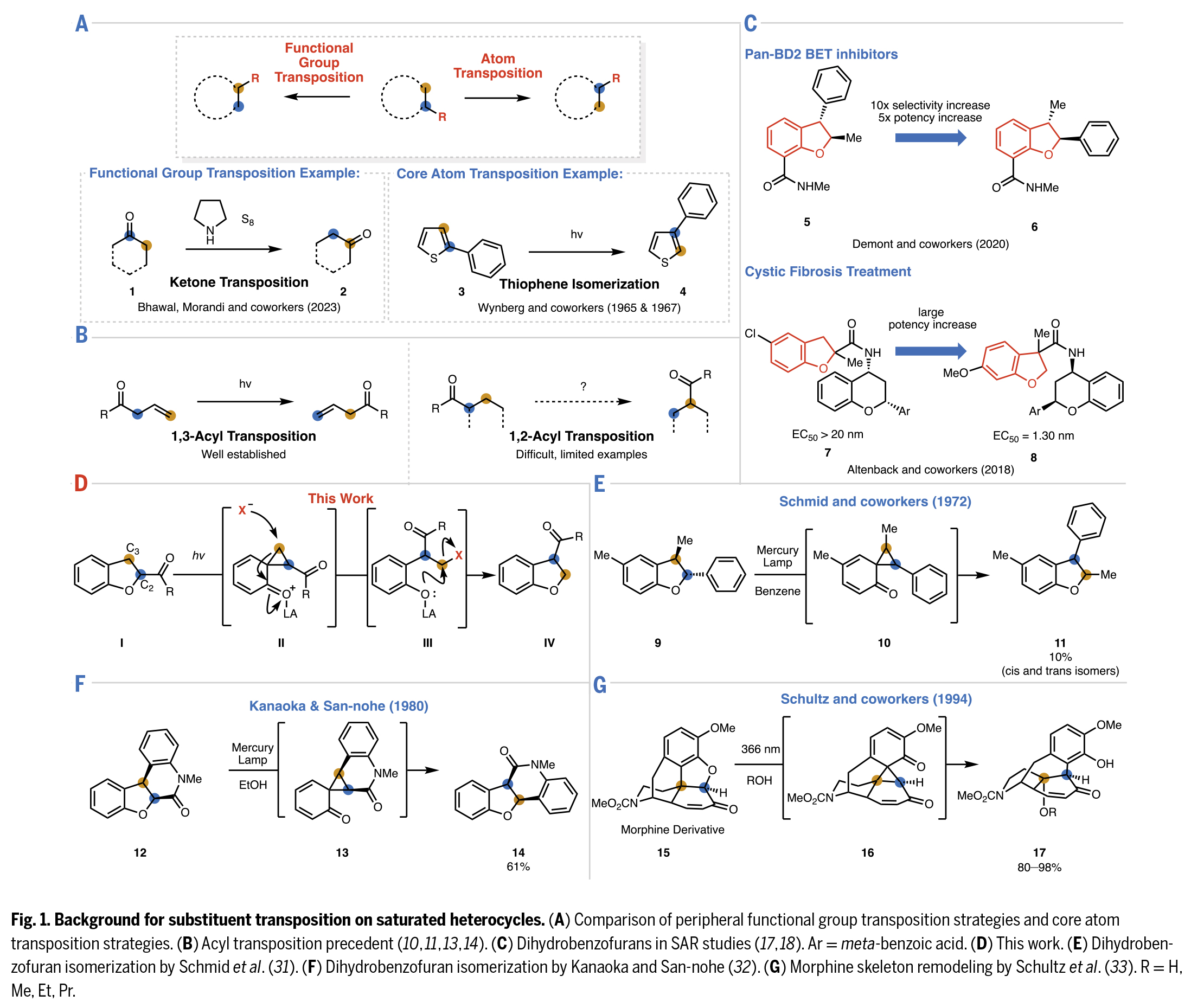
饱和杂环化合物是药物分子中常见的骨架结构。在药物发现过程中,化学家们往往需要对官能团位置不同的类似分子进行大量筛选。因此,开发能够实现饱和杂环上现有取代基重排至周边位点的合成方法具有重要意义。近日,加州大学伯克利分校Richmond Sarpong课题组通过C2-酰化二氢苯并呋喃的C2-C3位光化学交换反应,实现了形式上的1,2-酰基转位。虽然该反应的净效应是使羰基取代基在碳环上移动一个位点,但其作用机制实际上是通过保持C-CO键不变的同时交换骨架原子来实现的。该反应以二氢苯并呋喃为底物,其过程可能涉及光化学途径生成的环丙烷中间体(Fig. 1)。欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
(图片来源:Science)
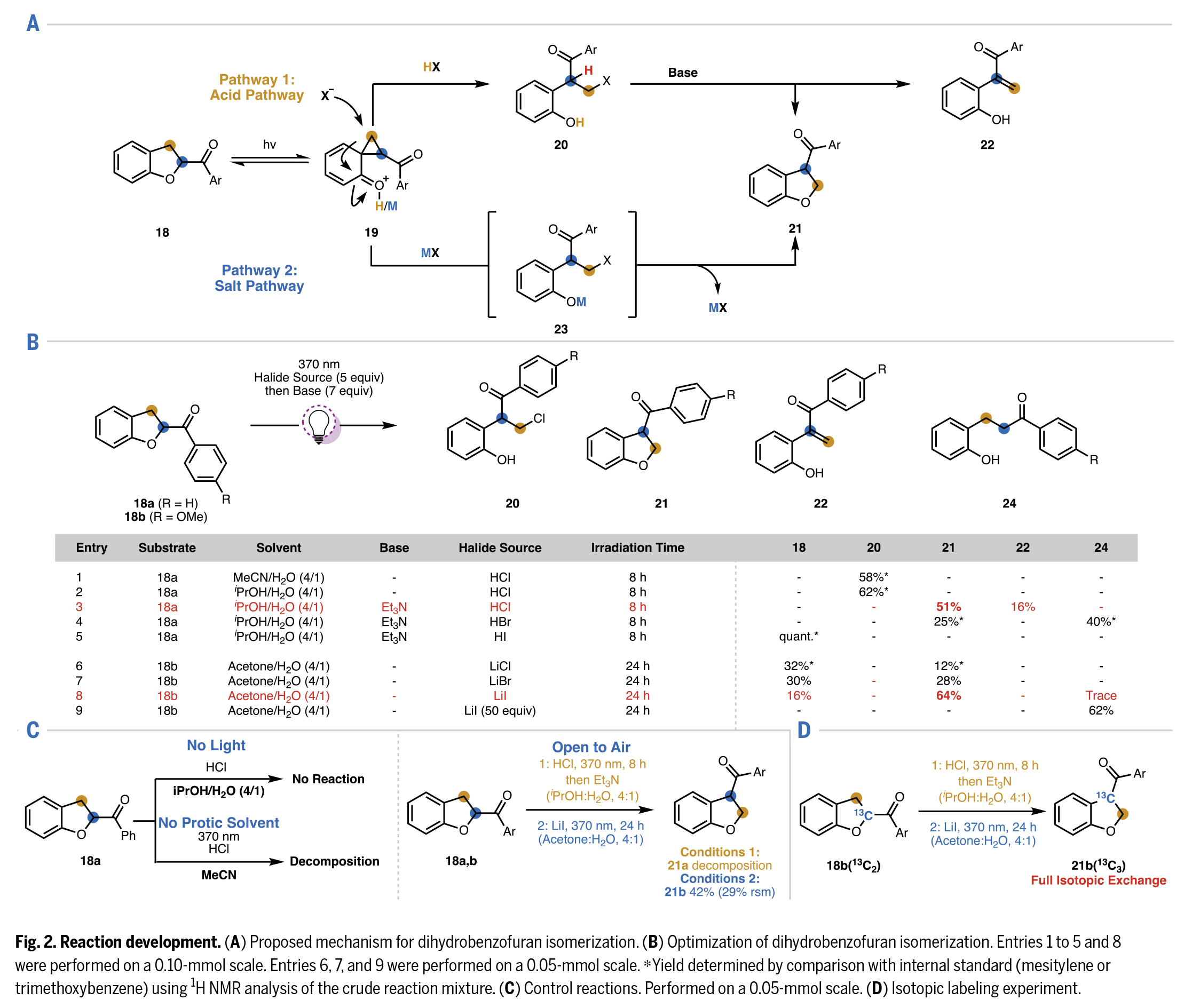
首先,作者观察到在氮气保护的极性质子溶剂中,模板底物18在稀盐酸溶液里经370 nm的紫外光照射后,可发生预期的氯化反应生成产物20。该产物即使在持续光照下也不会进一步反应(Fig.2B)。对照实验证实,该反应需要光照条件、质子溶剂和无氧环境(Fig.2C)。随后,作者经过大量条件筛选,确定异丙醇与水的4:1混合体系为合成伯氯代物20的最佳溶剂。随后作者通过对多种碱的筛选得出三乙胺能在有效促进目标环化反应的同时,最大限度地减少22的生成。考虑到若使用不同卤化氢可能避免这一竞争性消除反应,作者考察了其他卤化酸。当使用HBr时,未观察到消除反应产生的烯酮副产物,但由于单电子转移过程导致开环还原产物24a的生成(收率40%),使得总收率下降。而使用HI时反应完全被抑制,原料定量回收。最终确定HCl是最适宜的酸催化剂。
作为对初始发现的补充,作者提出一个假设,即无需采用两步一锅法,卤化物源可直接引发异构化反应。进一步筛选表明,反应在丙酮:H2O(4:1)混合溶剂中光照24小时效果最佳。相较于LiCl或LiBr,亲核性更强的LiI可获得更高收率(Fig.2B)。值得注意的是,使用LiI的酰基转位反应可在敞口空气条件下进行,收率仅有轻微的下降(Fig.2C)。
鉴于HCl和LiI条件各有优势,作者开发了两种二氢苯并呋喃底物形式芳基酮(芳酰基)转位方案:条件1为两步一锅法(370 nm光照8小时+HCl,随后用Et3N处理);条件2为一步法(LiI光照24小时)。此外,作者通过同位素标记实验(18b(13C2)转化为21b(13C3))验证了所提出的骨架重排机制。核磁共振(NMR)分析证实在两种优化条件下,同位素标记的碳原子均在产物的C3位(Fig.2D)。
(图片来源:Science)
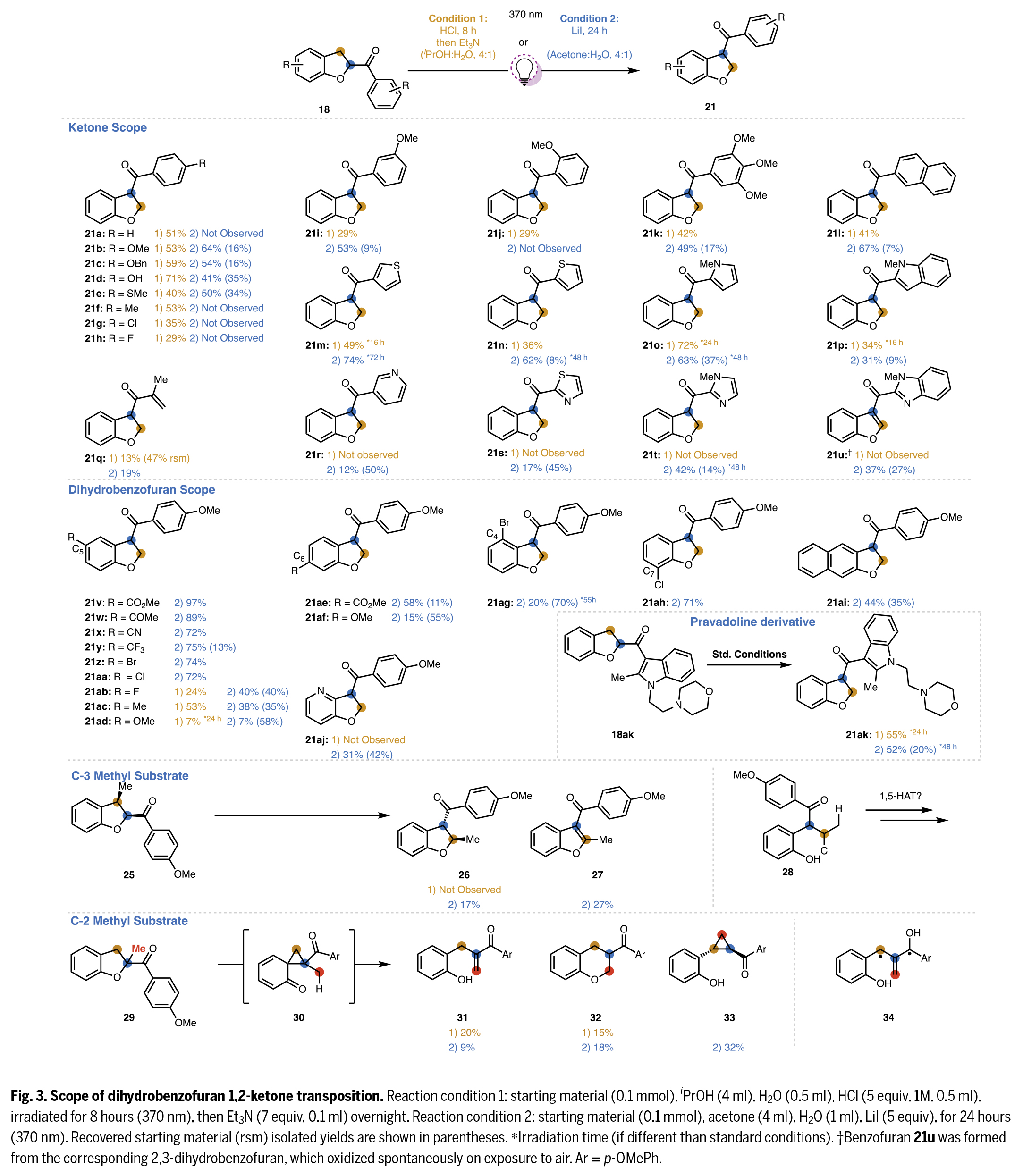
在得到了最优反应条件后,作者对此转化的底物范围进行了探索(Fig. 3)。实验结果表明该转化其具有广谱的底物范围。一系列不同取代的酮和二氢苯并呋喃均具有良好的兼容性,以中等至良好的产率得到相应的产物21a-21aj。值得注意的是,Pravadoline衍生物同样可以兼容,证明了此转化的实用性。
为考察C3位取代基对转位反应的影响,作者研究了带有额外取代基底物的反应兼容性。采用LiI条件时,顺式C3-甲基二氢苯并呋喃25成功转位生成反式产物26及芳构化产物27。尽管不能完全排除顺式26生成后发生芳构化或分解的可能性,但实验未检测到任何顺式产物。而条件1仅导致分解反应,这可能是因为开链氯代加合物28会进行不利的Norrish II型反应,而刚性结构26则不会发生此类副反应。
在两种条件下光照2-甲基底物29均未获得目标产物。该反应仅以低收率生成烯酮31、32和环丙烷33,这些产物可通过制备薄层色谱与其他微量副产物有效分离。推测31是由30通过形式[1,5]-σ迁移重排产生,而31经6-endo-trig氧杂-迈克尔加成生成32。另一种途径是31的进一步激发可能促使苄位氢原子通过1,4-氢原子转移至激发态芳酮,形成的中间体34,该中间体通过自由基重组生成环丙烷33。
(图片来源:Science)
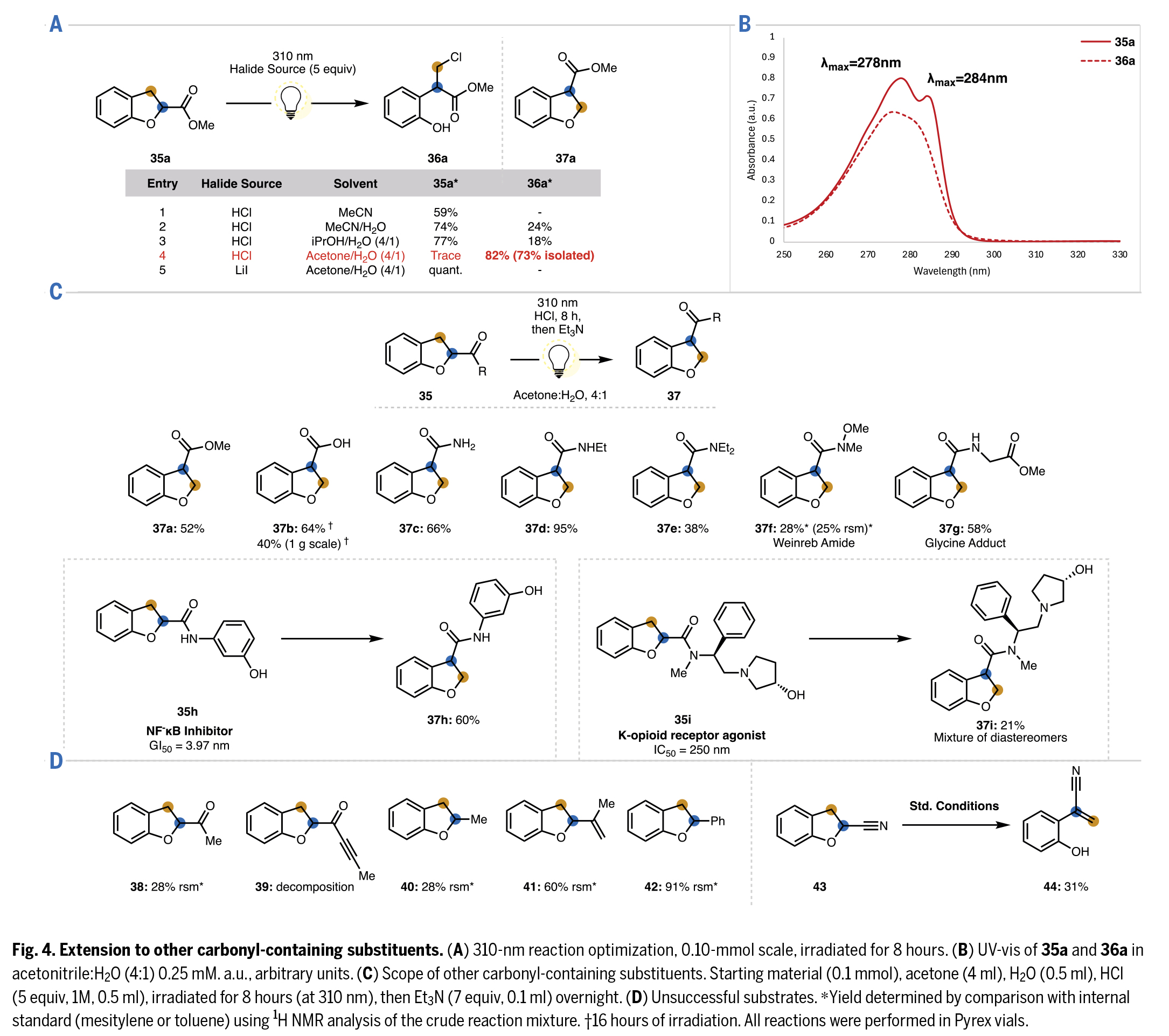
受前人关于二氢苯并呋喃在紫外光作用下形成螺环丙烷的报道启发,作者研究了其他类型含羰基官能团的转位反应,特别是那些在370 nm反应条件下不具有光反应活性的基团(Fig. 4)。实验发现,通过中心波长为310 nm的光照,甲基酯35a能以高产率转化为伯氯代物36a(Fig.4A)。在丙酮:水(4:1)混合溶剂中反应收率最高(entry 4)。由于酯基的酸性低于先前研究的芳基酮,所得伯卤代物(36a)能有效转化为转位产物(37a),且不会发生竞争性的卤素消除副反应。此外,作者尝试使用LiI直接将35a转化为37a未获成功(entry 5)。
随后,作者对C2-酯基取代的二氢苯并呋喃及其相应氯代加合物(35a和36a)进行了紫外-可见吸收光谱测定(Fig.4B)。值得注意的是,35a和36a在300 nm以上波长区域基本无显著吸收(Pyrex玻璃瓶可滤除300 nm以下波长)。虽然紫外-可见光谱显示35a在300 nm以上波长应该不能有效吸光并发生光化学反应,但光化学反应效率往往在比底物最大吸收波长更长的波段更高。此外,丙酮在300-330 nm区间有微弱吸收,可能通过溶剂光敏化作用促进反应。特别值得注意的是,35a在λmax = 284 nm处的特征吸收峰在氯代加合物36a中消失,这合理解释了为何36a在反应条件下能保持光稳定性。
其他含羰基官能团—包括羧酸(35b)和酰胺类(35c-35g)—均能在310 nm光照下成功实现转位(Fig.4C)。即使不稳定的Weinreb酰胺35f也能耐受反应条件,其N-O键未发生均裂,最终获得转位产物与原料的混合物。C3-羧酸35b的转位反应在克级规模仍能进行,仅收率略有下降,证明该方法在高度可衍生化结构单元上的可放大性。这种对多种含羰基官能团的普适性显著提升了该反应在生物活性分子合成中的应用价值。然而,烷基酮38和炔基酮39在所发展转位条件下均发生分解(Fig.4D)。
(图片来源:Science)
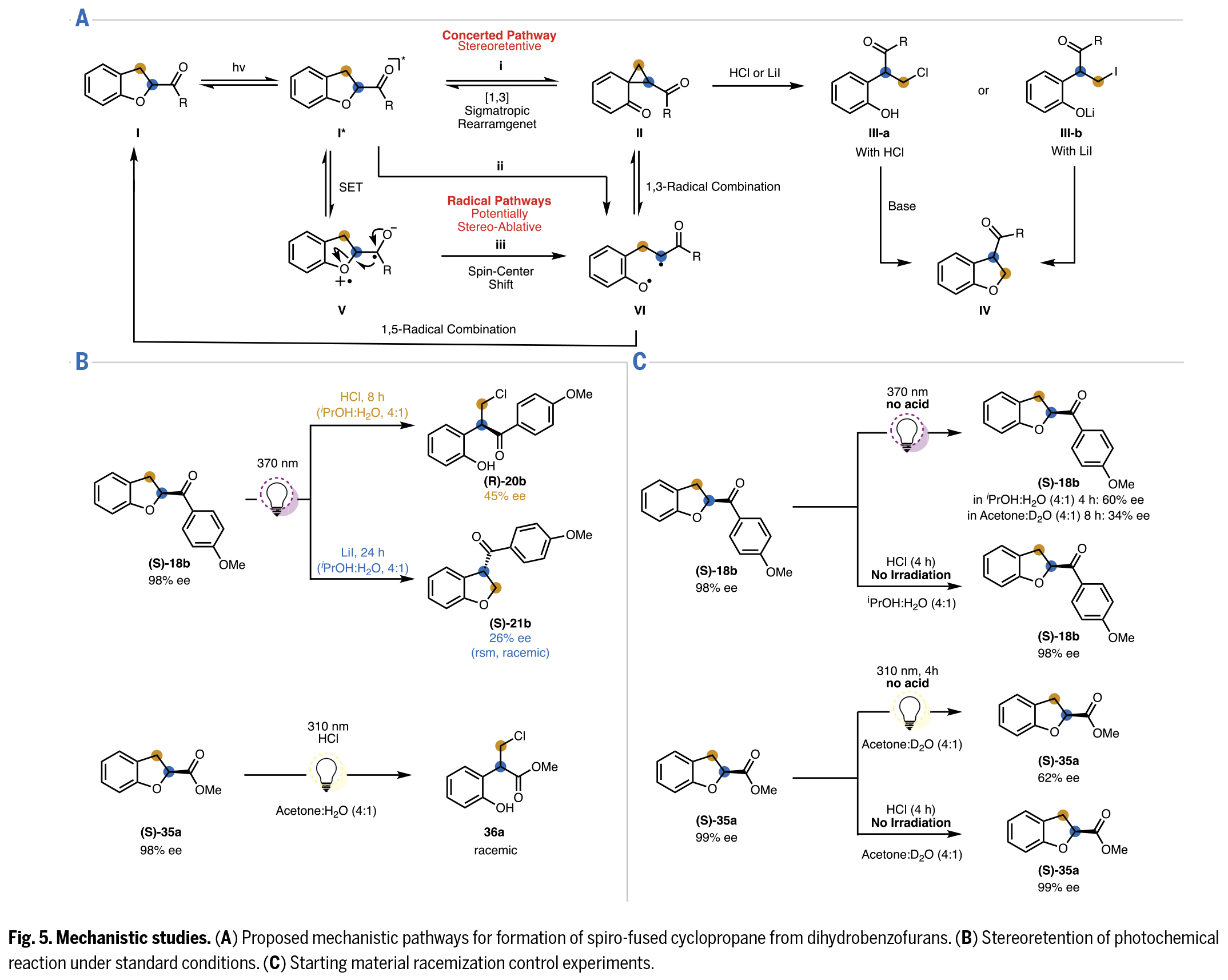
最后,作者提出了光化学激发态下关键螺环丙烷中间体形成的三种可能路径:(i)[1,3]-σ迁移重排;(ii)C-O键均裂后发生1,3-双自由基重组;(iii)电子转移伴随自旋中心转移开环,继而发生双自由基重组(Fig.5A)。在某些反应中所表现出的立体保持特性支持了立体专一性的σ迁移重排机制;而从类似二氢呋喃底物的光化学异构化研究中分离出的双自由基产物及其寿命测定结果为双自由基中间体的存在提供了证据。此外,环丙烷中间体也是通过双自由基机制形成的。
作为研究的重要组成部分,作者对手性富集起始原料产物的立体保留(或消旋)特性进行了定量分析。实验观测显示,当使用高对映体纯度的底物时,无论是条件1还是条件2都会导致产物对映选择性显著降低(Fig.5B)。虽然这一现象初步支持了通过双自由基中间体导致消旋化的机理,但也不能排除另一种可能性。即手性富集的螺环丙烷中间体(II)可能通过热力学或光化学驱动的杂乙烯基环丙烷重排转化为起始原料,且该过程在某些情况下已被证实会通过导致消旋的自由基机理进行。
对照实验证实,回收原料的消旋化源于光照作用而非酸性添加剂(Fig.5C)。在丙酮:D2O体系中开展的补充实验显示,回收的消旋化原料未检测到氘代现象,这排除了通过光致烯醇化导致消旋的路径。这些结果表明消旋过程是通过形成双自由基中间体(VI),随后发生1,5-位重组重新生成I而实现的(Fig.5A)。
(图片来源:Science)
总结
Richmond Sarpong课题组开发了一种通过光化学转化生成螺环丙烷中间体、实现二氢苯并呋喃C2-C3位酰基取代基形式转位的合成策略。该策略利用非常规的骨架重排反应,解决了传统方法难以实现的周边官能团位置迁移难题。这种通过光化学途径获取去芳构化螺环丙烷中间体的策略,将为二氢苯并呋喃骨架的多样化修饰提供新思路。机理研究表明,螺环丙烷中间体的形成部分涉及自由基反应机制。
文献详情:
1,2-Acyltransposition throughphotochemical skeletalrearrangement of2,3-dihydrobenzofurans.
Ryan T. Steele, Motohiro Fujiu, Richmond Sarpong*.
Science,2025, 388, 631-638.
DOI:10.1126/science.adv9915
来源: 化学加
