
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
近日,美国普林斯顿大学David W.C. MacMillan、加州大学伯克利分校Jeffrey R. Long与美国默克公司Chi “Chip” Le团队报道了一种氮杂环卡宾(NHC)介导的脱氧反应与镍催化芳基羧酸脱羧反应的协同策略,成功实现了C(sp³)−C(sp²)键的构建。同时,通过对多种芳基-烷基交叉偶联产物的高效合成以及对复杂分子(如药物分子、天然产物及生物分子)后期衍生化,进一步证明了反应的实用性。文章链接DOI:10.1021/jacs.4c15827
图片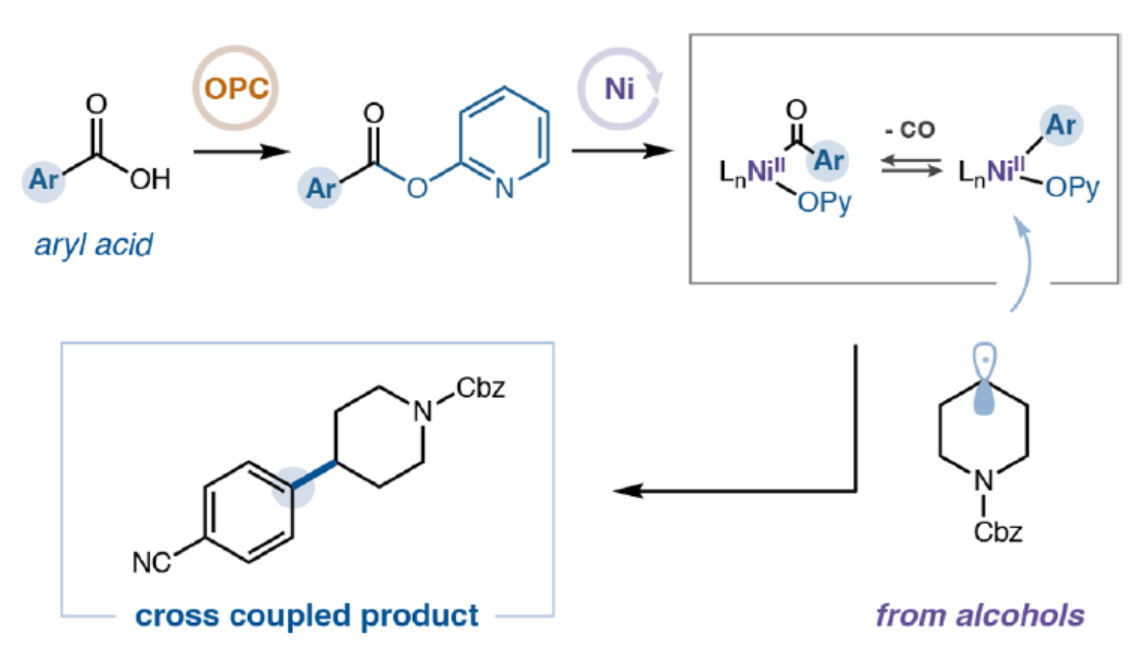
(图片来源:J. Am. Chem. Soc.)
正文
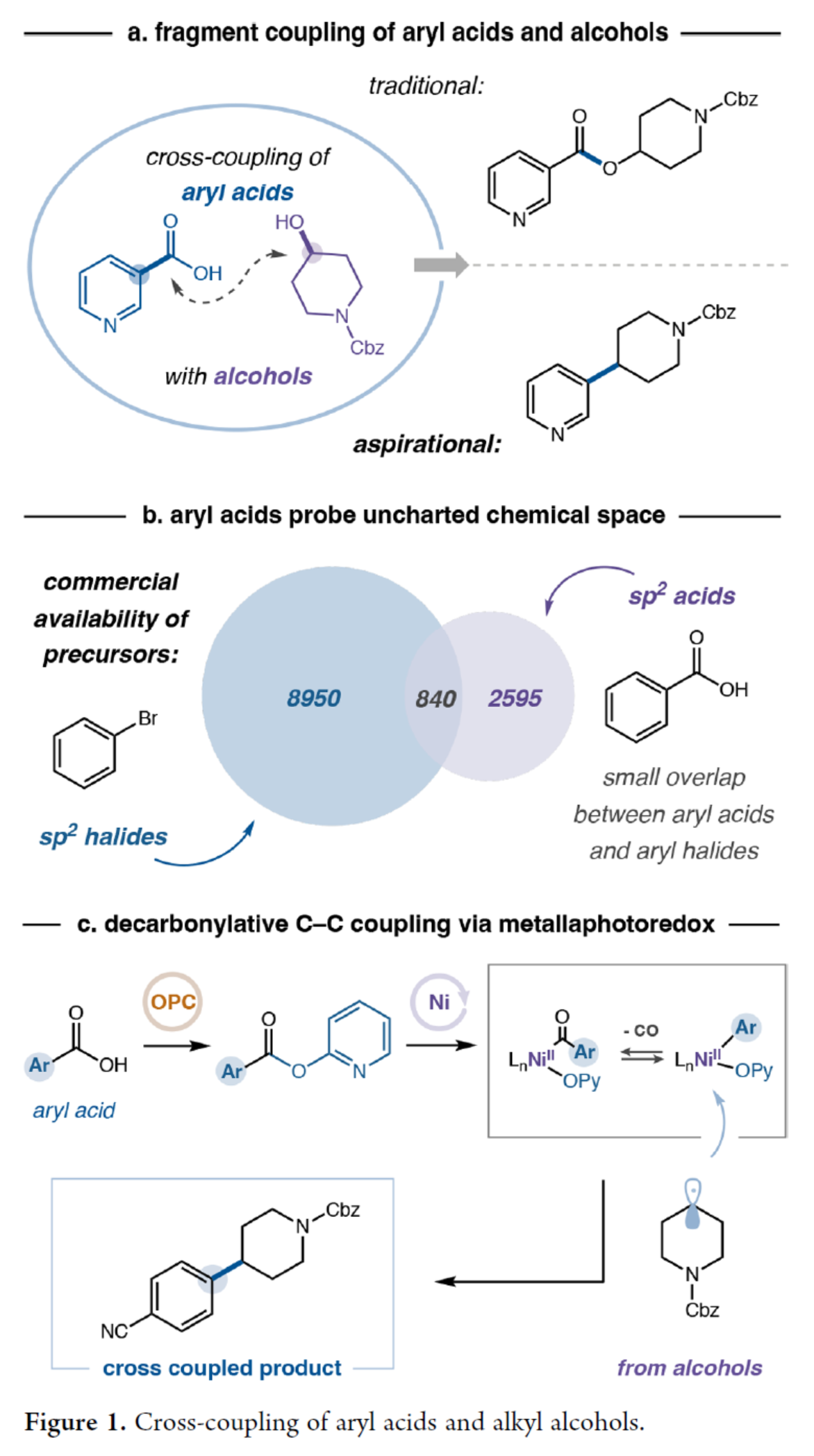
C(sp³)−C(sp²)结构单元在药物研发中具有特殊重要性,其可通过富含C(sp³)的分子骨架赋予三维空间复杂性,进而在药物设计与发现中发挥关键作用。同时,此类骨架能够显著提升化合物的溶解性、生物利用度及药代动力学性质,因而在药物分子开发中备受青睐。传统上,C(sp³)−C(sp²)键的合成主要依赖于过渡金属(如钯)催化的交叉偶联方法,需使用烷基/芳基有机金属试剂与卤代物反应。然而,由于该策略对昂贵或难以获取的起始原料与催化剂的依赖性,限制了其广泛应用。近年来,镍催化领域的进展为还原型C(sp³)−C(sp²)偶联开辟了新途径——该方法与钯催化体系形成互补,并可在更温和条件下实现更广泛的底物兼容性。尽管取得了这些进展,现有方法仍主要依赖芳基与烷基卤化物作为反应组分,但此类原料常面临成本高昂、制备困难或官能团耐受性有限等问题。为此,MacMillan等团队致力于寻求一种可替代的交叉偶联组分,并最终选定烷基醇与芳基羧酸作为理想候选——因其兼具商品化易得性、结构多样性及合成普适性等优势。虽然醇和芳基羧酸通常会发生酯化反应,但MacMillan等团队旨在探究金属光氧化还原策略是否能够促进C(sp³)−C(sp²)键的脱羰基化形成(Figure 1a)。此外,芳基羧酸所探索的化学空间通常是芳基卤化物尚未涉足的领域。因此,该方法将提供互补的反应活性,并拓展进入一个大部分未被探索的化学空间的途径(Figure 1b)。最近,Weix和Cernak课题组通过镍催化脱羰基反应实现了碳-碳键的构建,这一研究展示了芳基羧酸作为多功能合成子参与交叉偶联反应的潜力。受此启发,近日,MacMillan等团队开发了一种将醇脱氧与芳基羧酸活化相结合策略,成功构建了一个能够高效形成C(sp³)−C(sp²)键的催化体系,并具有优异的官能团耐受性(Figure 1c)。欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
(图片来源:J. Am. Chem. Soc.)
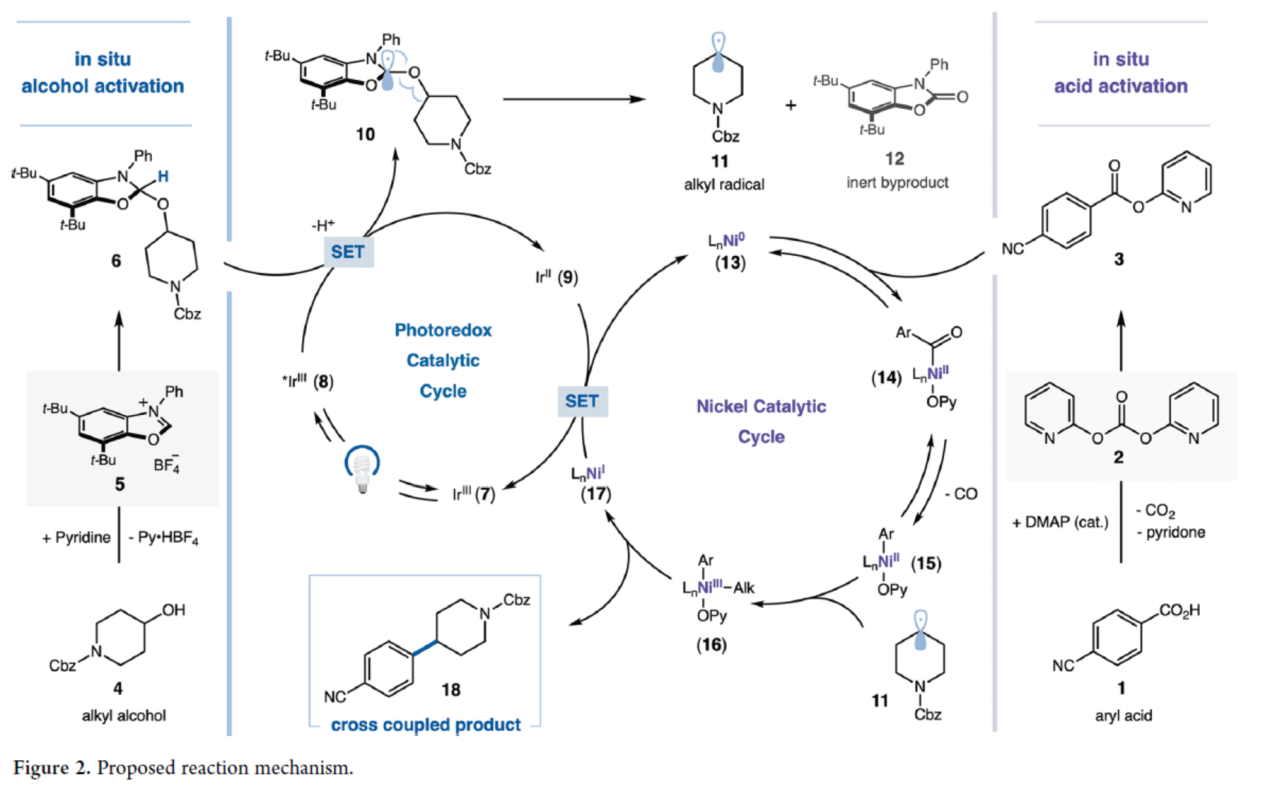
首先,作者提出了一种合理的双重镍/光氧化还原催化交叉偶联反应的机理(Figure 2)。最初,将芳基羧酸(1)与二吡啶-2-基碳酸酯(DPC,2)及催化量的DMAP预混合,无需纯化即可原位生成活化酸(3)。在另一反应容器中,醇底物(4)与NHC盐(5)在弱碱性条件下反应,同样无需进一步纯化即可原位生成活化NHC-醇加合物(6)。其次,在可见光激发光催化剂Ir[dF(CF3)-ppy]₂(dtbbpy)PF₆(7)时,会生成具有长寿命氧化性质的三重激发态配合物(8),其可通过NHC-醇加合物(6)的单电子转移(SET)过程发生还原淬灭,从而形成还原态的Ir(II)光催化剂(9)与中间体(10)。随后,中间体(10)的快速去质子化及β-断裂过程将释放出惰性芳构化副产物(12),并生成烷基自由基(11)。同时,作者推测,Ni(0)配合物(13)对活化酸(3)的氧化加成将生成Ni(II)配合物(14),其经历脱羰反应后生成中间体(15)。烷基自由基(11)可被镍催化剂快速捕获,形成Ni(III)-烷基中间体(16),其通过还原消除后生成目标交叉偶联产物(18),并释放出Ni(I)中间体(17)。最终,Ir(II)配合物(9)与Ni(I)配合物(17)之间经单电子转移(SET)过程将Ni(I)还原为Ni(0),同时使Ir(II)被氧化,从而同步完成光氧化还原循环和镍催化循环。
(图片来源:J. Am. Chem. Soc.)
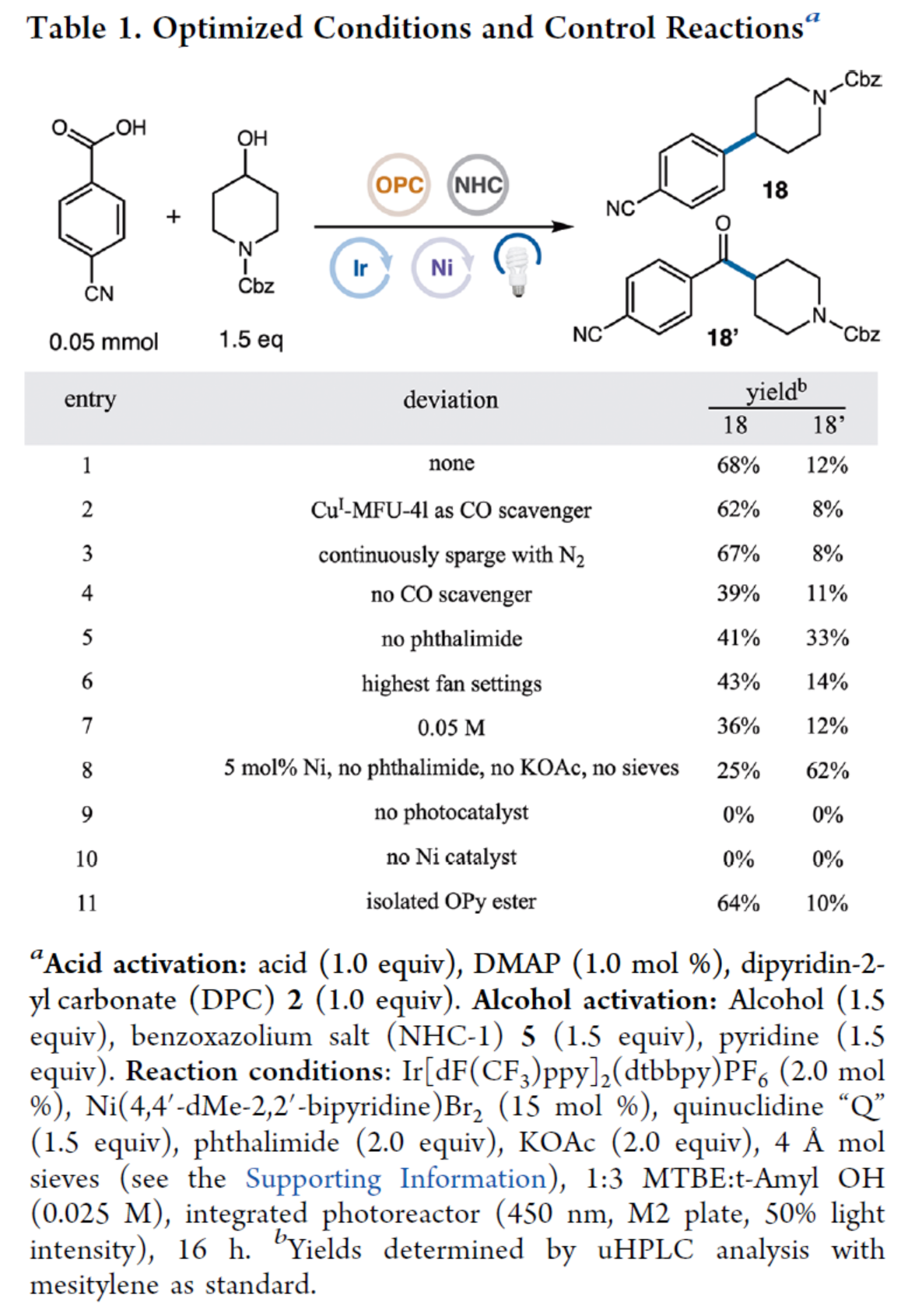
其次,作者以4-氰基苯甲酸与醇衍生物作为模型底物,进行了相关反应条件的筛选(Table 1)。酸化合物活化的条件为:即羧酸化合物(1.0 equiv)与二吡啶-2-基碳酸酯(DPC,2)(1.0 equiv)在DMAP(1.0 mol %)催化下反应。醇化合物活化的条件为:即醇化合物(1.5 equiv)与吡啶(1.5 equiv)在NHC-1(5,1.5 equiv)催化下反应。在活化两种底物后,以Ir[dF(CF3)ppy]2(dtbbpy)PF6(2.0 mol%)作为光催化剂,Ni(4,4’-dMe-2,2’-bipyridine)Br2(15 mol%)作为金属催化剂,奎宁环“Q”(1.5 equiv)作为配体,邻苯二甲酰亚胺(2.0 equiv)作为添加剂,KOAc(2.0 equiv)作为碱,4 Å作为添加剂,蓝色LEDs作为光源,在MTBE:t-Amyl OH(比例为1:3)混合溶剂中反应,可以68%的收率得到偶联产物18以及12%收率的酮副产物18’。
(图片来源:J. Am. Chem. Soc.)
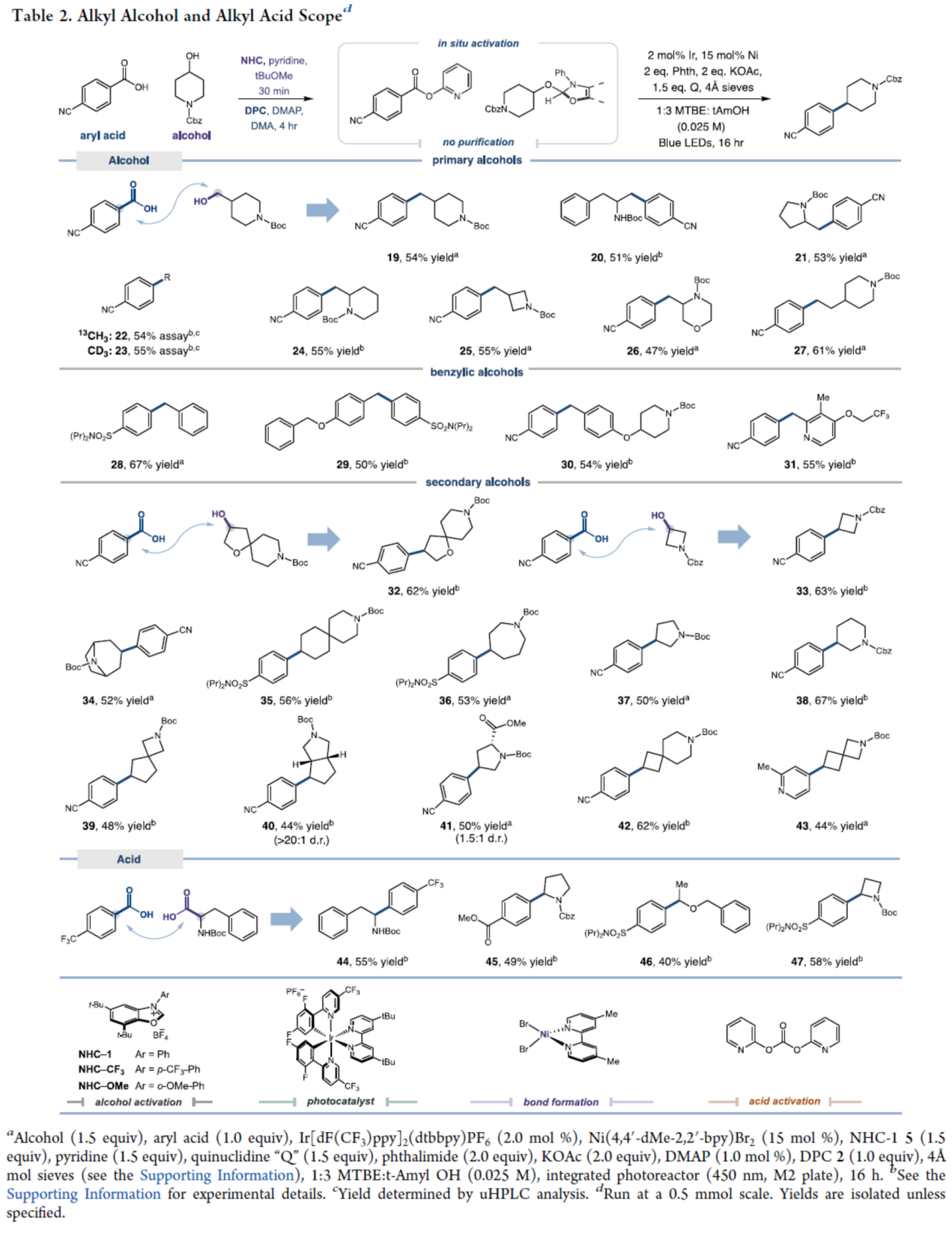
在获得上述最佳反应条件后,作者对烷基醇与烷基羧酸的底物范围进行了扩展(Table 2)。首先,一系列一级醇与二级醇,均可顺利进行反应,获得相应的产物19-43,收率为44-67%。其中,通过市售甲醇即可简便地引入富氢和富碳同位素单元,从而避免了冗长的从头合成步骤(如22和23)。其次,羧酸也可作为烷基自由基,其可从丰富的α-氨基酸原料氧化生成,如苯丙氨酸(44,收率55%)、脯氨酸(45,收率49%)、α-羟基酸(46,收率40%)和非经典α-氨基酸氮杂环丁烷(47,收率58%)中生成,并获得相应的C(sp³)−C(sp²)偶联产物。
(图片来源:J. Am. Chem. Soc.)
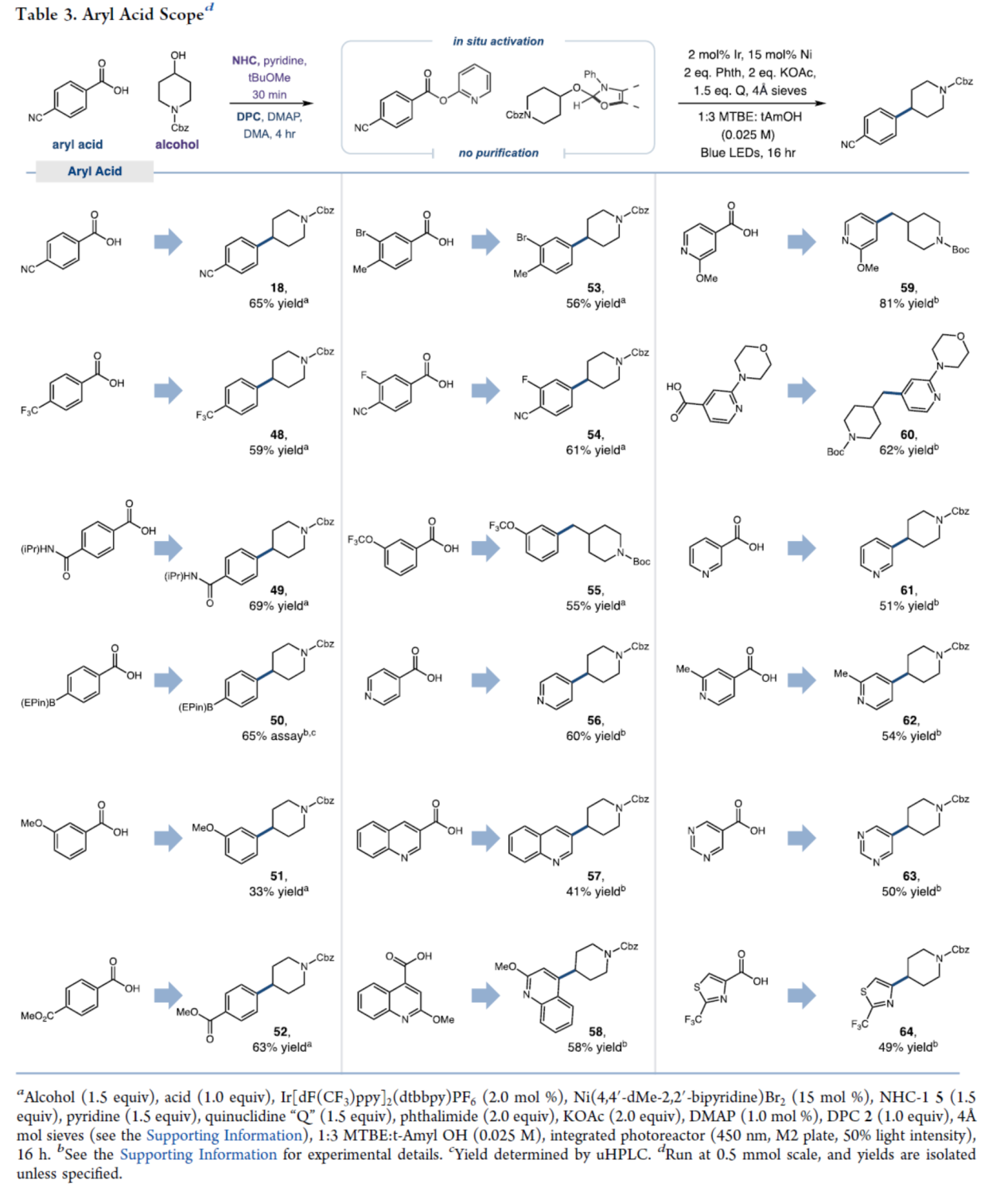
紧接着,作者对芳基羧酸的底物范围进行了扩展(Table 3)。首先,具有不同电性和位阻的苯甲酸,均可顺利进行反应,获得相应的产物18和48-55,收率为33-69%。其次,衍生自吡啶、喹啉、嘧啶、噻唑等杂芳基羧酸,也与体系兼容,获得相应的产物56-64,收率为41-81%。值得注意的是,一系列活性基团,如硼基、烷氧羰基、卤素等,均与体系兼容。同时,由于2-吡啶酯与镍催化剂的氧化加成速率较慢,同时伴随脱羰反应速率的下降,导致富电子的羧酸底物(如51)收率有所降低。
(图片来源:J. Am. Chem. Soc.)
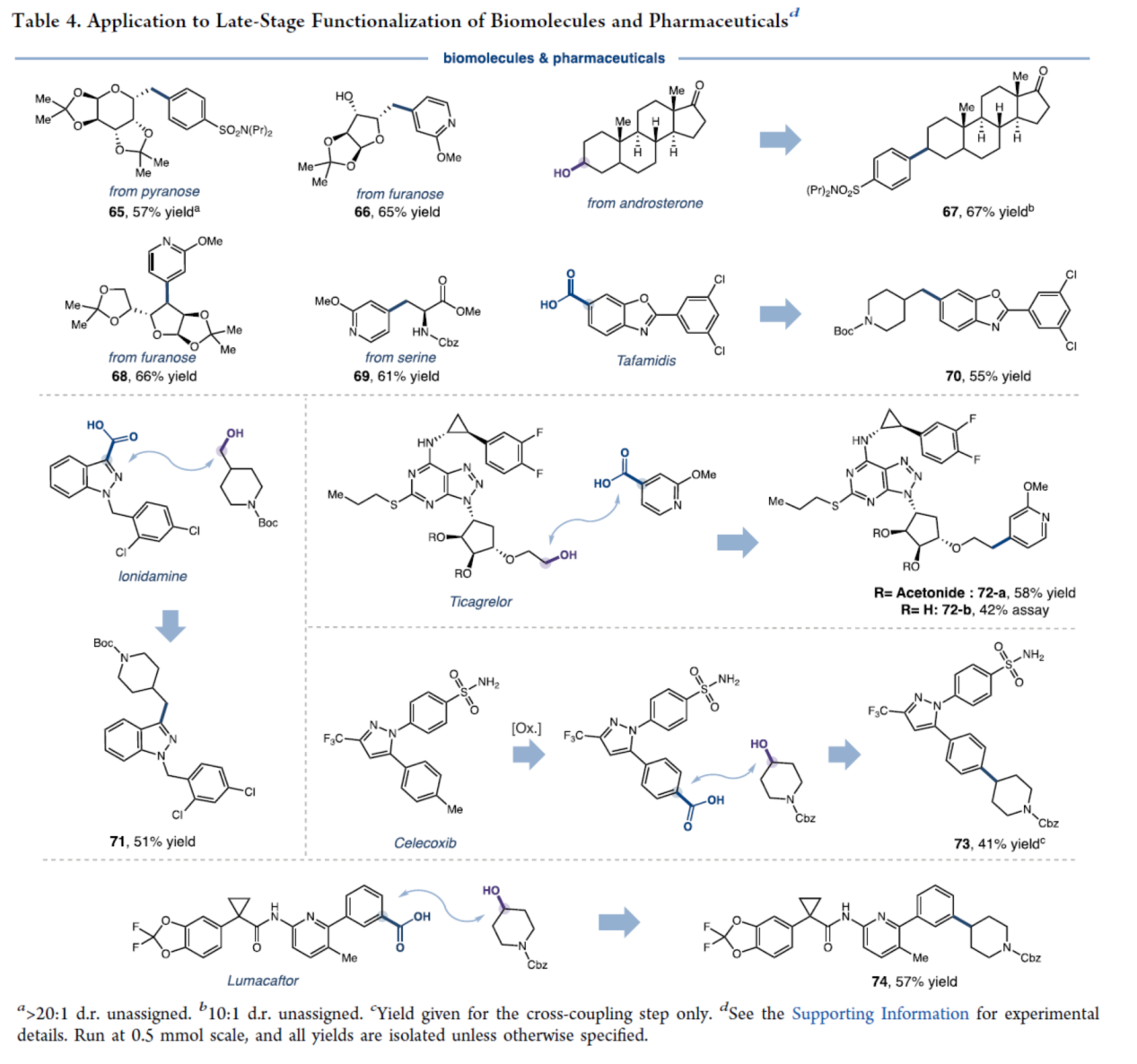
最后,作者对反应的实用性进行了研究(Table 4)。首先,该策略可用于一系列药物与生物分子的后期衍生化,如氯苯唑酸(Tafamidis)、氯尼达明(Lonidamine)、鲁玛卡托(Lumacaftor)、替格瑞洛(Ticagrelor)、糖类化合物、α-氨基酸丝氨酸的衍生物、睾酮(Androsterone)等,获得相应的化合物65-74,收率为41-67%。值得关注的是,在呋喃糖(66)与替格瑞洛(72-b)的反应中,观察到显著的区域选择性,即在二级醇共存条件下,一级醇位点被专一性活化。
(图片来源:J. Am. Chem. Soc.)
总结
David W.C. MacMillan等人报道了一种芳基羧酸与烷基醇的形式交叉偶联策略,实现了C(sp³)−C(sp²)键的构建。该方法不仅为传统交叉偶联技术提供了一种互补策略,也为传统的酯化反应提供了一个正交替代方案。其次,通过将NHC介导的脱氧过程与镍催化脱羰成键机理相结合,作者开发了一种适用于广泛脂肪醇和芳基羧酸的双重镍/光氧化还原催化方法。这种转化的关键因素是加入分子筛和其他清除剂,能够有效去除CO,从而最大限度地减少酮类副产物的生成,并提高反应的转化率。此外,使用非传统的交叉偶联底物,如醇和芳基羧酸,这种方法可以开辟构建富含C(sp3)的新化学空间。
文献详情:
Aryl Acid-Alcohol Cross-Coupling: C(sp3)−C(sp2) Bond Formationfrom Nontraditional Precursors.
Eva Lin, Johnny Z. Wang, Edna Mao, Stephanie Tsang, Kurtis M. Carsch, Cesar N. Prieto Kullmer,Ryan E. McNamee, Jeffrey R. Long,* Chi “Chip” Le,* David W.C. MacMillan*.
J. Am. Chem. Soc.2025
DOI:10.1021/jacs.4c15827
来源: 化学加
