
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 中华医学会
中华医学会 
作者:韩冰 北京协和医院 主任医师
审核:江茜 中国人民解放军总医院第三医学中心 主任医师
在医学的广阔领域中,罕见病以其独特的发病机制和临床表现,始终吸引着科学家和临床医生的关注。先天性纯红细胞再生障碍性贫血(DBA)便是这样一种罕见而复杂的遗传性疾病,主要影响婴幼儿,以纯红细胞减少和特定的临床特征为标志。
该病得名于1938年首次报道该病症的两位外国学者Diamond和Blackfan,即Diamond-Blackfan Anemia(DBA)。该病的主要特征是红细胞生成障碍导致的贫血,而白细胞和血小板则相对正常。
DBA的病因深藏于遗传物质之中,主要由基因突变所致。具体而言,这些突变影响了线粒体内血红蛋白合成过程中关键结构蛋白的功能,使得血红蛋白前体成分无法正常合成,进而引发贫血。
值得注意的是,约30%-50%的DBA病例存在家族遗传模式,多为常染色体显性或隐性遗传,其余病例可能因新发突变引起。RPS19基因突变是最常见的变异,约占所有病例的一半,该基因位于第19号染色体短臂和8号染色体短臂上,编码的核糖体蛋白对红细胞成熟至关重要。
此外,环境因素也在DBA的发病过程中扮演了一定角色。虽然具体的环境因素尚未完全明确,但可以推测,某些外部条件可能加剧或触发基因突变的表达,进而增加患病风险。
DBA的临床表现颇具特征性。首先,该病起病较早,90%的患者在一岁以内便会出现症状,表现为贫血、网织红细胞计数低以及骨髓中红系细胞缺乏。与之相比,患者的白细胞和血小板数量基本正常,这使得DBA在血液系统疾病中显得尤为独特。
此外,患者可能伴有特殊面容、颈蹼、斜视、生长发育畸形以及心脏和肾脏等内脏畸形。值得注意的是,相较于普通人群,DBA患者成年后罹患肿瘤的风险显著增高。
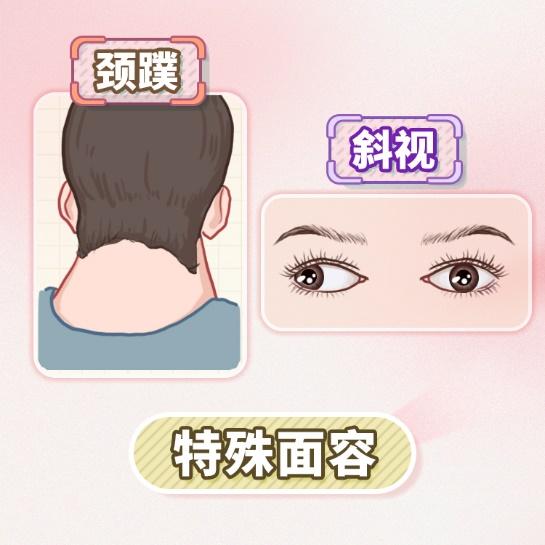
图1 原创版权图片,不授权转载
另外,DBA不仅影响儿童的生长发育和器官功能,还可能导致铁过载。因为贫血会使DBA患者反复进行输血,而反复输血会使得外来红细胞在体内分解后释放的铁无法有效排出,积累在肝脏、心脏和内分泌腺体中,引起“黑肝”现象,即脏器在核磁共振图像中呈现黑色。并且,DBA本身导致铁调节系统紊乱,机体错误地认为体内缺铁,从而加快铁吸收,进一步导致铁过载。铁过载会增加肝硬化、肝癌风险,引发心衰、甲状腺功能低下、早发性糖尿病以及性功能障碍等问题。
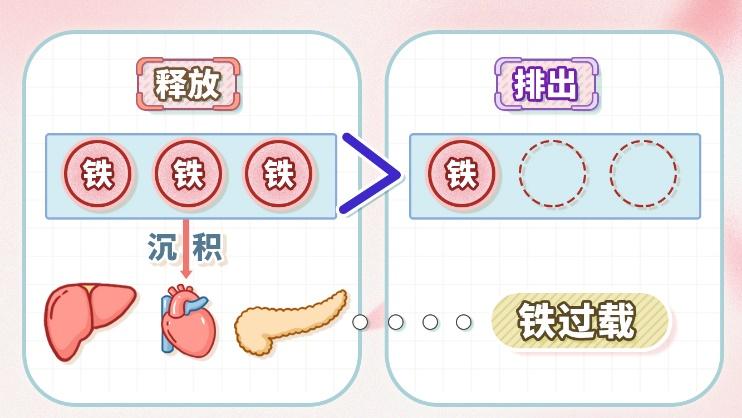
图2 原创版权图片,不授权转载
在诊断DBA时,全面的临床评估和实验室检查是必不可少的。医生通常会通过血常规、骨髓穿刺、染色体及基因检测等手段,综合评估患者的病情。特别是基因检测,能够直接检测到RPS19等关键基因的突变情况,为DBA的确诊提供有力依据。
与其他疾病如一过性红细胞减少、Pearson综合征、范可尼贫血和先天性角化不良相比,DBA具有独特的贫血表型。例如,Pearson综合征患者骨髓中可见环形铁粒幼细胞,而范可尼贫血和先天性角化不良的基因表现与DBA明显不同。
尽管DBA难以预防,但父母在常规健康筛查中发现贫血时,不应轻易下结论,而需保持高度关注,及时复诊,并在孕期避免感染和不良环境因素。对于已知家族中有DBA的夫妇,应考虑遗传咨询,评估再次生育的患病风险。产前检查虽不能确保发现DBA,但定期监测胎儿发育和器官状况,以及出生后的血红蛋白监测和必要时的基因检测,有助于早期识别疾病迹象。

来源: 中华医学会
内容资源由项目单位提供
