
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
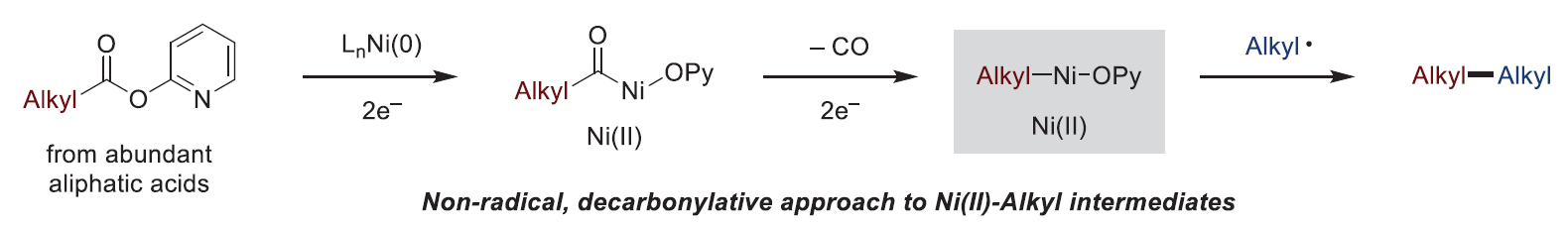
近日,美国威斯康星大学麦迪逊分校(University of Wisconsin–Madison) Daniel J. Weix课题组发展了一种非自由基策略,实现了烷基羧酸酯的氧化加成和去羰基化过程,合成了稳定的单烷基镍(II)中间体。实现此过程的关键是配体双(4-甲基吡唑基)吡啶的使用,其可以有效加速去羰基化过程,稳定烷基镍(II)中间体,并使非循环的镍(0)羰基物种变得不稳定。该反应的反应性在纯自由基方法难以实现的C(sp3)-C(sp3)成键反应中得到了证明,其可以选择性的使一级羧酸酯与一级烷基碘化物发生偶联。文章链接为DOI:10.1126/science.abi4860。
(图片来源:Science)
正文
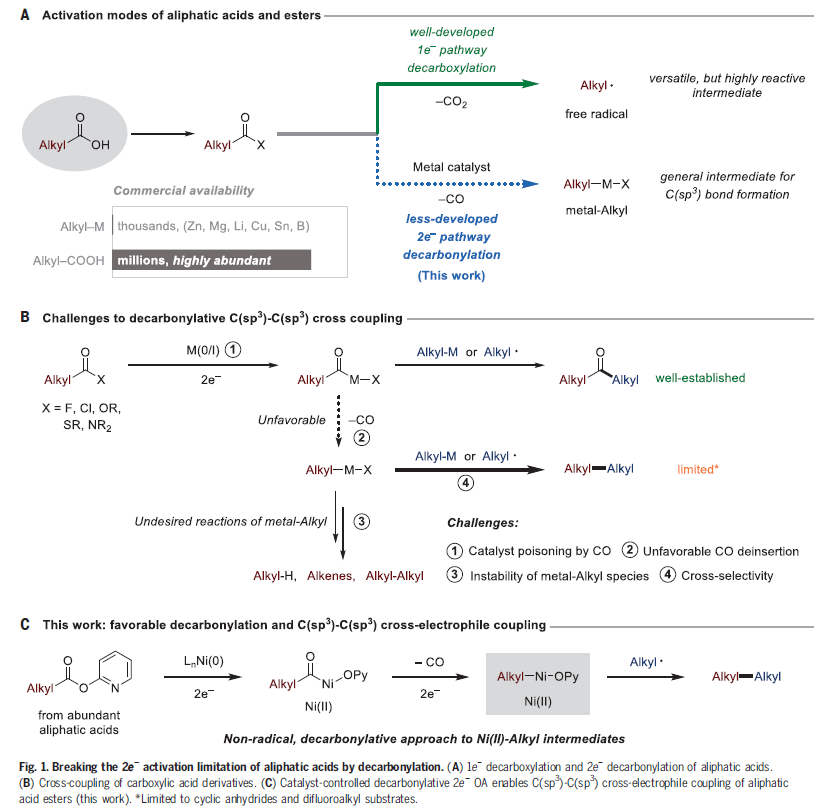
众多镍催化的交叉偶联反应依赖于有机镍中间体的形成,但由于单烷基镍物种难以形成的局限性限制了镍催化C(sp3)交叉偶联反应的发展。如果能够实现从来源丰富的羧酸酯中得到单烷基镍(II)物种将具有重要的应用价值。但在镍催化的偶联反应中,羧酸衍生物主要通过脱羧形成烷基自由基,因此不具备此类反应性。最近,美国威斯康星大学麦迪逊分校Daniel J. Weix课题组发展了一种非自由基反应策略,实现了烷基羧酸酯的氧化加成和去羰基化过程,实现了稳定的烷基镍(II)络合物的制备。值得注意的是,利用此策略可有效实现一级羧酸酯与一级烷基碘化物的交叉偶联,从而构建C(sp3)-C(sp3)键(Fig. 1)。
(图片来源:Science)
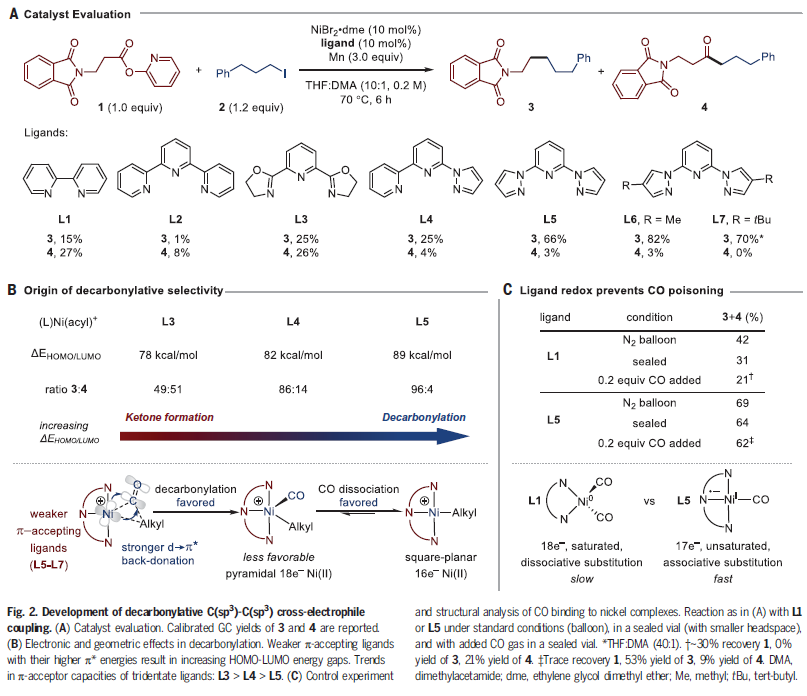
首先,作者以羧酸酯1和烷基碘化物2作为模板底物对反应中的配体进行了筛选(Fig. 2)。
通过对一系列配体筛选,作者得出Mebpp L6 (1 mol%)为最佳,该配体通过对吡唑上的电性进行调控,可以以82%的产率得到脱羰偶联产物3,且通过气相色谱法测得副产物酮4的产率仅为3%。其中L6的刚性三齿骨架结构可以增强CO的释放能力,这主要是由于(L6)NiII(alkyl)(CO)+是18电子络合物,但镍(II)在方形平面的16电子构型中更稳定。DFT计算表明,(L5)Ni(CO)是一种具有开放配位位点的方形平面络合物,且其基态为(L5•–)NiI(CO)。此计算结果与实验结果相一致,L5与L1的反应相比受CO中毒的影响较小。上述实验结果表明,三齿配体可以避免CO中毒,这与可逆生物氧化还原反应、光解或化学计量试剂是互补的。
(图片来源:Science)
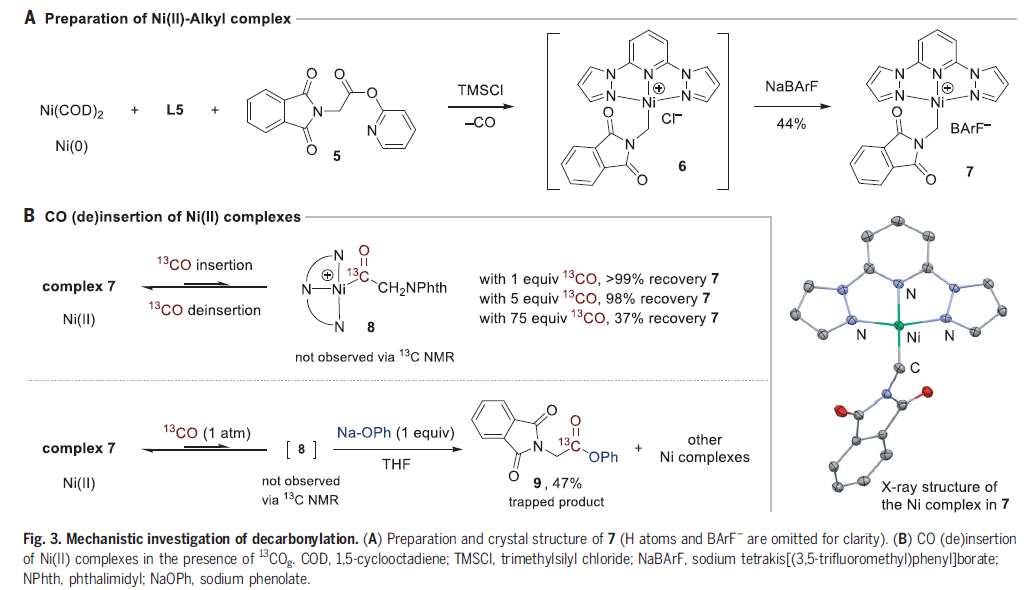
为了更好的理解羰基步骤以及交叉选择性的起源,作者进行了机理研究(Fig. 3)。首先,作者在室温下将等摩尔的Ni0(COD)2、L5和邻苯二甲酰亚胺羧酸吡啶酯5混合在四氢呋喃(THF)中来探索其脱羰过程(Fig. 3A)。由于吡啶酮配体会使得烷基镍配合物的分离存在一定的困难,因此作者使用TMSCl将其交换为Cl-,最后交换为BArF-后分离出稳定的、带正电荷的烷基镍(II)络合物7,并对其进行了包括单晶X-射线衍射在内的全面表征。镍的配位环境为扭曲的正方形平面结构,与相关的(L3)NiII(Ar)+络合物的配位环境相似。由于作者能够通过中间氯代络合物中间体分离出7,因此这一结果表明吡啶酮配体在脱羰基步骤中发挥了作用。
接下来,为了更好的了解脱羰步骤,作者测试了络合物7与CO的反应性。尽管CO配位或插入在热力学上有利于烷基镍(II)配合物的生成,但作者无法通过核磁共振直接观察到插入后的产物8(Fig. 3B)。加入13COg(1 equiv)后仍然是不配位的游离CO,且在~280 ~ ~200 ppm之间作者没有观察到羰基物种或酮类副产物的峰。只有在75 equiv的13COg的存在下,7的浓度才会下降,但得到的是未知产物。当7用13COg (1atm)和亲核试剂(NaOPh, 1 equiv)捕获时,可以以47%的产率得到13C标记的酯产物9(Fig. 3C)。这些结果表明[(bpp)NiII(CH2NPhth)]+7比插入的络合物8更具热力学稳定性,但这些络合物在动力学上是可获得的。
(图片来源:Science)
考虑到自由基脱羰的先例以及烷基酯与镍催化脱羰交叉偶联的罕见性,作者寻求进一步的证据来证明脱羰是此催化反应的主要途径(Fig. 4)。1e-脱羰途径的显著特征是游离烷基自由基的生成。而当使用α-手性羧酸作为底物时,会导致立体化学的消失。相比之下,脱羰是一个1,1-迁移过程,其是立体特异性的。为了阐明关键的脱羰步骤,作者考察了N-Cbz-4-(叔丁硅氧基)-脯氨酸酯的化学计量和催化脱羰氘化(Fig. 4A)。羟脯氨酸衍生物的两种非对映体(10和12)分别反应形成氘化产物11和13,且具有近乎完全的立体特异性(分别>99%和98%的立体化学保持)。为了确保这些化学计量结果与催化作用相关,作者随后在d1-2,4,6-三甲基苯甲酸(14)存在的情况下(作为烷基镍捕获剂),将10与烷基碘2进行了标准的脱羰交叉亲电偶联。虽然由于β-氢消除存在使得立体定向的交叉偶联产物产率很低,但反应中的烷基中间体可以被14氘化生成11,产率为63%,并完全保留了立体化学。这些结果与通过自由基途径得到的偶联产物的非对映体比(dr)为1:1的结果形成了鲜明的对比。
接下来,作者研究了烷基镍(II)络合物7对烷基碘化物2的反应性和选择性(Fig. 4B)。虽然烷基镍(II)络合合物在构建C(sp3)-C(sp3)键中常被提及,但在化学计量反应中却很少使用。7与3-苯基-1-碘丙烷的化学计量反应可以生成交叉产物15,在添加或不添加还原剂的情况下产率分别为61%和54%。反应中并没有形成很多的二聚体产物16,这与文献报道的结果相一致,表明三齿烷基镍(II)络合物比双齿配体更能抵抗烷基配体歧化和/或自偶联。此外,1与环丙基碘甲烷反应只生成重排化合物17,符合1e-自由基机理(Fig. 4C)。以1,1-二苯乙烯为自由基捕获剂的催化反应进一步表明,烷基碘化物通过自由基中间体参与反应。
基于上述实验结果,作者提出了此转化可能的反应机理(Fig. 4D)。2-吡啶基酯与(L6)Ni0 20的2e- 氧化加成形成酰基镍(II)物种21。虽然作者证明20是可获得的,但并不能排除另一种途径,即19与酯经历可逆反应形成(L6)NiIII(acyl)(py)(X),随后被Mn还原为21。
不管它是如何形成的,中间体21都可以通过阳离子物种22和23迅速脱羰形成络合物24。阳离子烷基镍(II)络合物24可以通过自由基捕获的方式捕获烷基自由基,形成镍(III)物种25。二烷基镍(III)络合物可以通过还原消除形成新的C(sp3)-C(sp3)键和镍(I)物种19。作者认为交叉选择性的产生类似于C(sp2)-C(sp3)交叉亲电偶联。这两种烷基亲电试剂的区别在于它们的活化方式不同,2-吡啶酯作为非自由基烷基亲电试剂的等价物,而烷基碘化物作为1e-烷基自由基源。2-吡啶酮配体可诱导快速脱羰过程发生。烷基镍(II) 中间体足够稳定,可以避免二聚化,并且能够捕获烷基自由基并发生还原消除。
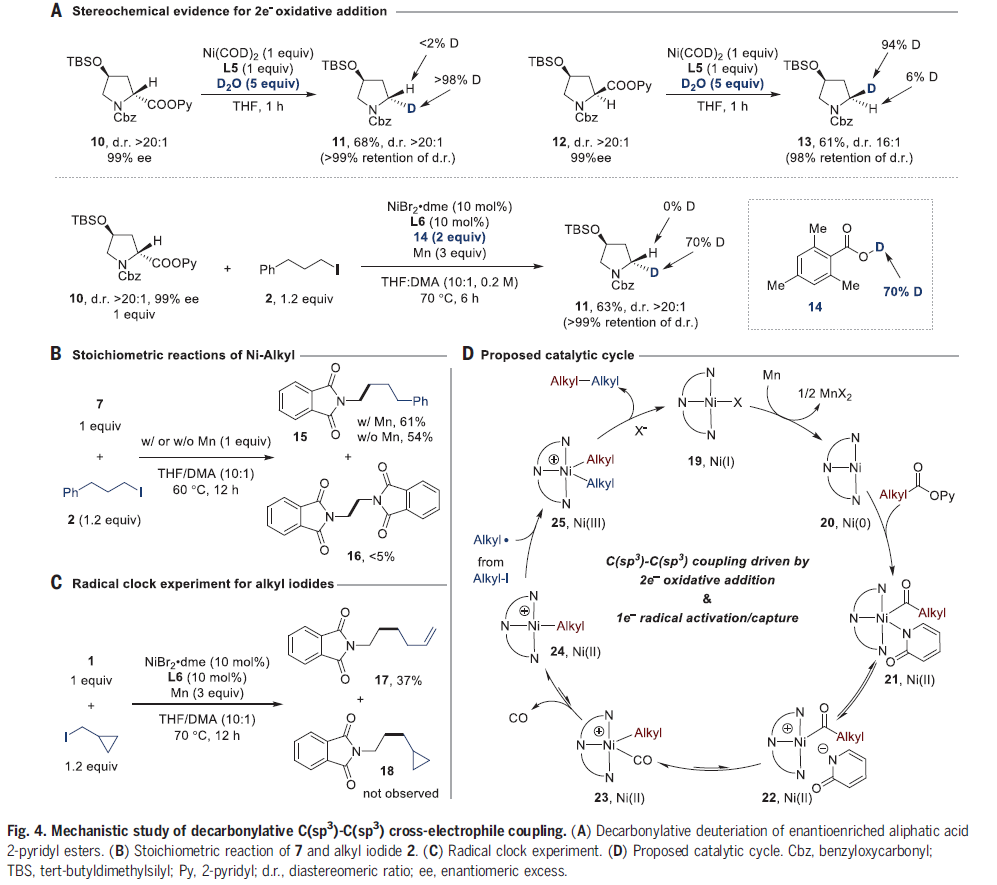
(图片来源:Science)
为了证明此策略的实用性,作者对底物范围进行了考察(Fig. 5)。实验结果表明一系列不同取代的烷基碘化物均可兼容,以27-75%的产率得到产物3-41。其中包括烷氧基、硅基、卤素、磺酰基、氰基以及酯基等均可兼容。值得注意的是,半乳糖、沙利度胺和Corey内酯衍生的烷基碘均可顺利实现转化,证明了此转化的实用性。此外,自然界来源丰富的脂肪羧酸同样具有良好的兼容性,包括甘氨酸、β-丙氨酸、高脯氨酸和γ-氨基丁酸2-吡啶基酯以及琥珀酸单酯均可实现转化,以30-77%的产率得到产物15,42-50。此外,复杂的天然产物同样可以兼容,以24-39%的产率得到产物51-54。
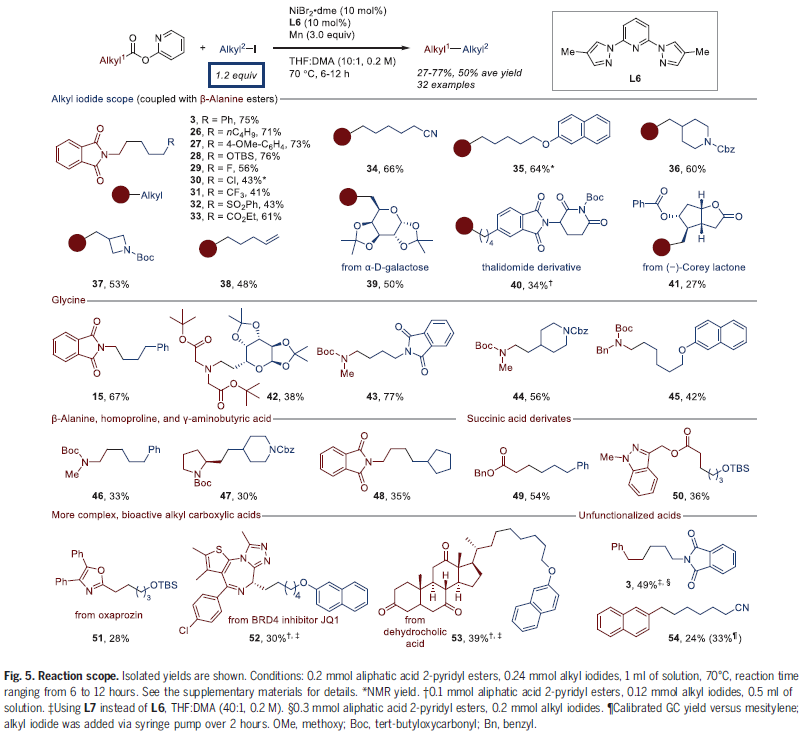
(图片来源:Science)
总结
Daniel J. Weix课题组发展了一种新的非自由基反应策略,由烷基羧酸酯的氧化加成和脱羰实现了单烷基镍(II)中间体的合成。三齿双(4-甲基吡唑)吡(Mebpp)配体的使用是实现此过程的关键,其可以加速脱羰过程并稳定烷基镍(II)中间体,使得选择性的C(sp3)-C(sp3)键的构建成为可能。此外,作者通过羧酸酯与烷基碘化物的偶联证明了此转化的实用性。
文献详情:
A decarbonylative approach to alkylnickel intermediates and C(sp3)-C(sp3) bond formation.
Zhidao Huang, Michelle E. Akana, Kyana M. Sanders, Daniel J. Weix*.
Science, 2024, 385,1331-1337.
https://www.science.org/doi/10.1126/science.abi4860.
来源: 化学加
