
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
近日,日本名古屋大学(Nagoya University)Kohsuke Ohmatsu和Takashi Ooi课题组报道了光催化磷叶立德的单电子氧化过程,实现了其与富电子烯烃和α,β-不饱和羰基化合物的连续组装,形成了一系列官能团化的六元碳环。这种三组分形式环加成过程包括连续的C-H官能团化和磷叶立德的Wittig反应,实现了一种卡拜类型的转化,包括将惰性的C-H和C=P键分别转化为C-C和C=C键。该反应可以作为一种强大的工具,实现从简单易得的底物快速构建多功能的合成砌块。相关成果发表在Nat. Synth.上,文章链接DOI:10.1038/s44160-024-00612-7。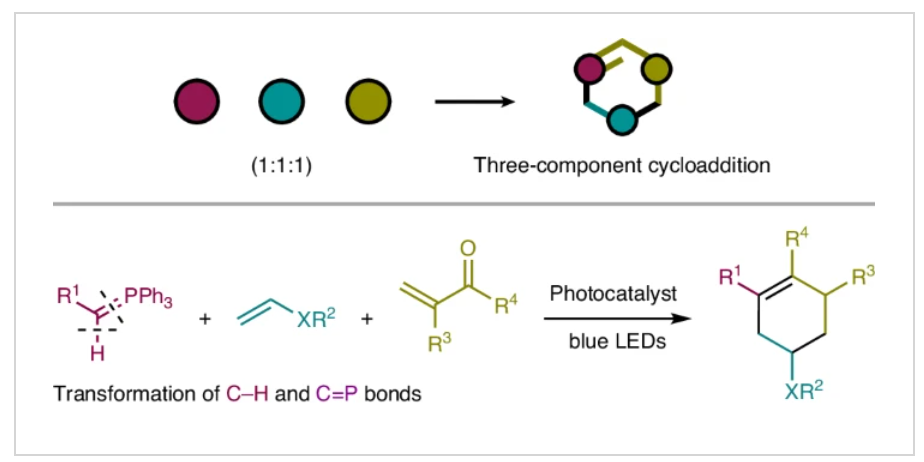
(图片来源:Nat. Synth.)
正文
单个碳原子上三价化学键的断裂可以生成具有自由基和卡宾反应活性的卡拜(Fig. 1a)。且该过程可以在一次反应操作中依次形成三根新的共价键,由此可以快速增加分子的复杂性。然而,由于这些极具活性的卡拜物种的产生和控制较为困难,因此到目前为止卡拜及其等价物的转化过程仅取得了有限的成功。现存的解决策略通常依赖于使用α-重氮离子(Y+C(=N2)Z;Y = I(III),R2S等,Z = 碳取代基),它们天生具有形成三根化学键的能力(Fig. 1b)。此外,新加坡南洋理工大学刘学伟课题组报道了单取代磷叶立德经历光氧化还原介导的C-H断裂形成叶立德自由基,其可以与烯烃进行自由基加成,随后与Hantzsch酯反应生成单烷基化产物(Fig. 1c)。这一反应过程证明了在光氧化还原催化下,利用磷叶立德的自由基反应性来实现极性反转的效果。然而,它们潜在的卡拜反应性,包括自由基和卡拜类型的反应性,还未得到充分利用。最近,日本名古屋大学Kohsuke Ohmatsu和Takashi Ooi课题组发展了光催化磷叶立德的单电子氧化过程,实现了与富电子烯烃和α,β-不饱和羰基化合物的连续加成,形成了一系列官能团化的六元碳环(Fig. 1d)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。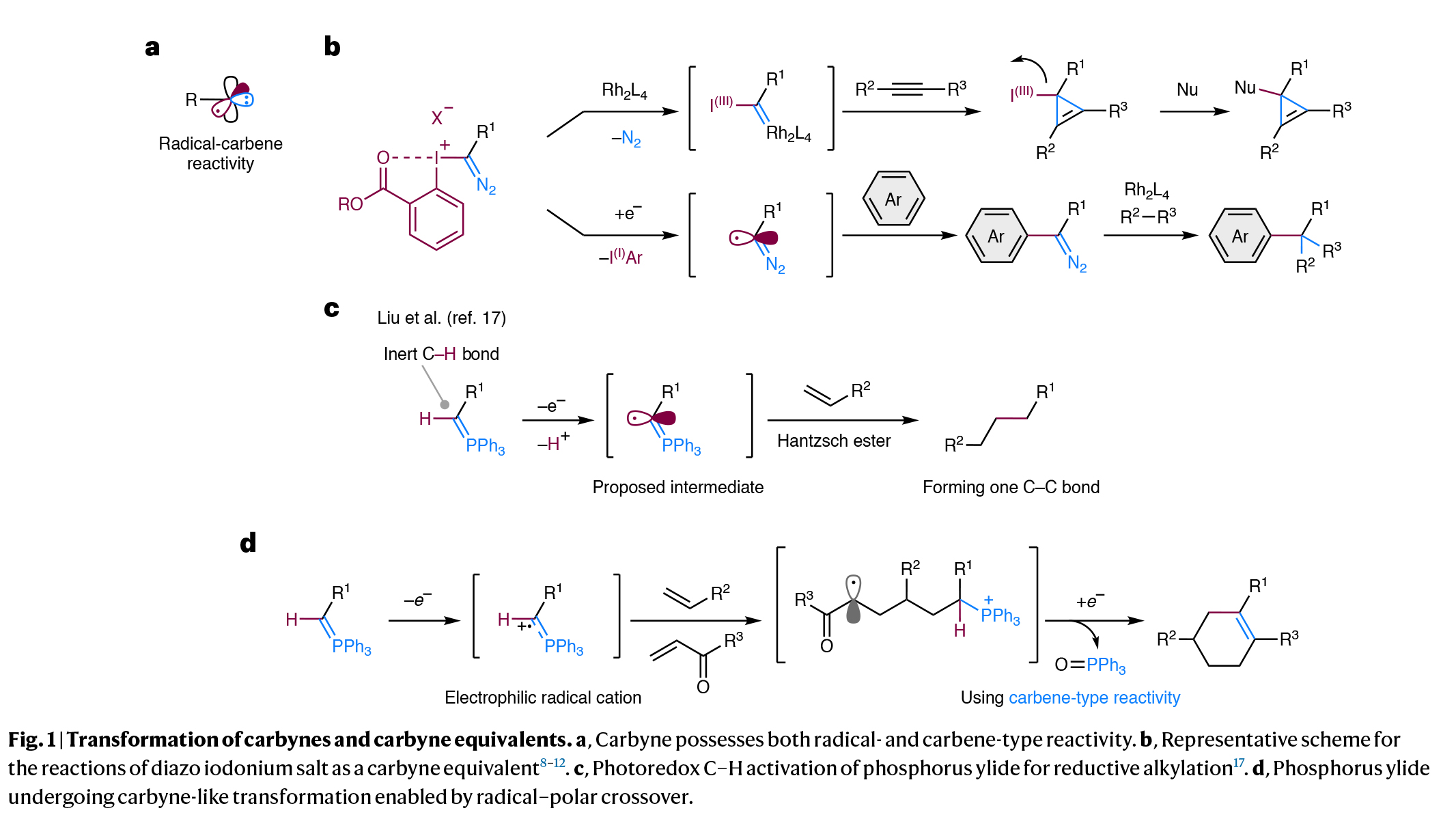
(图片来源:Nat. Synth.)
由于磷叶立德自身会与不饱和醛发生亲核加成,形成Wittig反应产物或Michael加合物,因此与富电子烯烃和高活性醛(如丙烯醛)的自由基-极性交叉反应具有一定的挑战性。因此,作者选择磷叶立德1a、叔丁基乙烯基醚和丙烯醛作为模板底物(Fig. 2)。由于1a的氧化电位为0.88 V,因此其在合适的激发态光催化剂存在下,可以很容易地通过单电子转移过程生成相应的自由基阳离子RC。作者认为RC具有足够的亲电性,可以与富电子的乙烯基醚反应,形成瞬态的亲核自由基中间体Int1-RC。随后将该自由基与丙烯醛进行共轭加成,得到α-甲酰基自由基中间体Int2-RC。由于与饱和甘汞电极相比,该物种的还原电位通常估计为-0.70 V,因此Int2-RC可以很容易地被PC的自由基阴离子还原为烯醇Int3。最后,通过与Int3的质子转移以及与Int4的分子内Wittig过程生成环加合物2a。
(图片来源:Nat. Synth.)
随后,作者对反应进行了尝试以及条件优化(Table 1)。实验结果表明,当使用1a(1.0 equiv),叔丁基乙烯基醚(1.0 equiv),丙烯醛(1.0 equiv),PC(3 mol%),在乙腈中,蓝光照射下,- 20 oC下反应15小时,可以以82%的核磁产率得到环加成产物2a(entry 6)。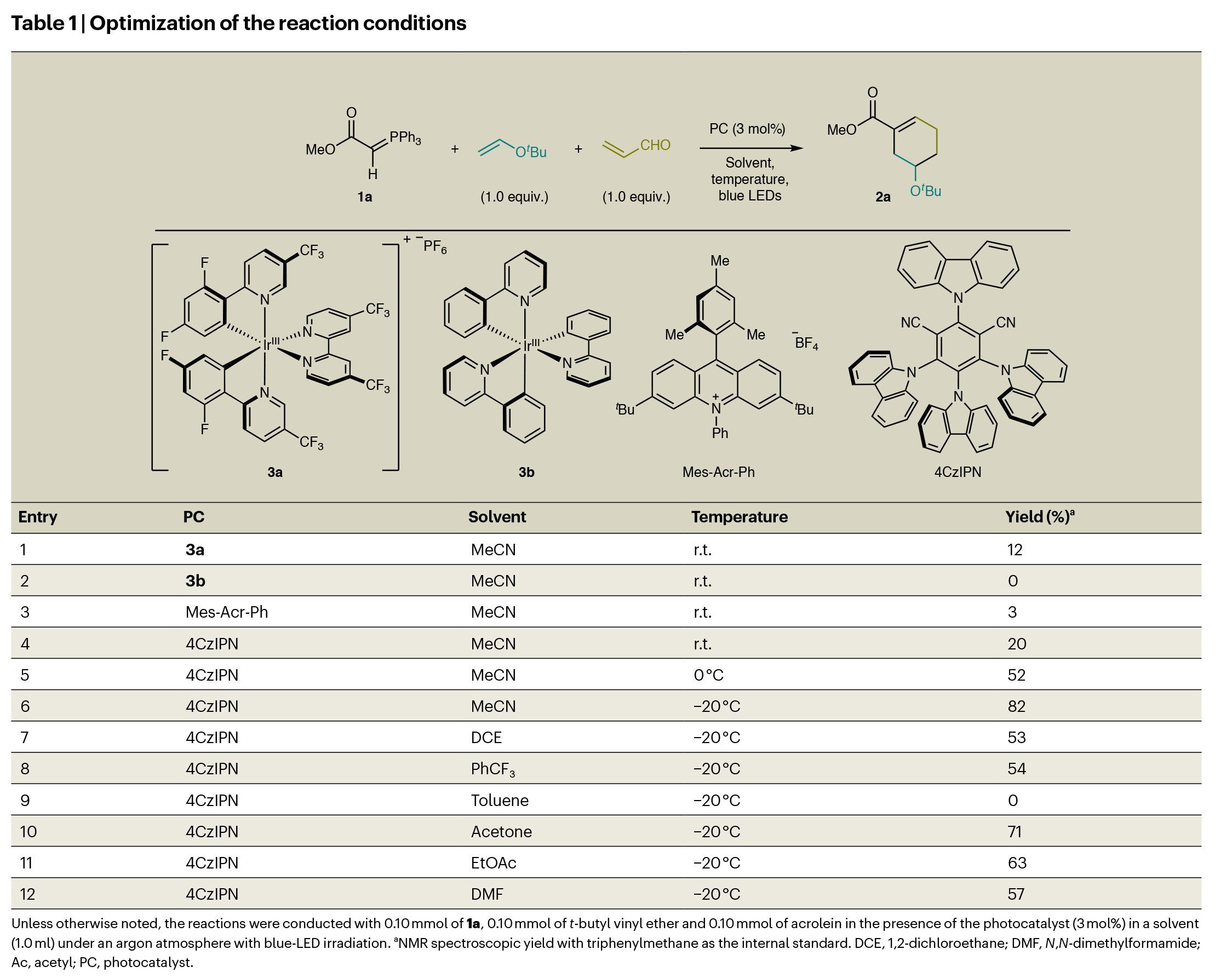
(图片来源:Nat. Synth.)
在得到了最有反应条件后,作者进行了底物范围考察(Fig. 3)。实验结果表明,一系列酯基取代的叶立德、酮和氰基取代的叶立德均可顺利参与反应,以59-84%的产率得到相应的环加合物2a-2g。此外,一系列不同的富电子烯烃,如烷基烯基醚、醛衍生的烯醇硅醚等均可实现转化,以40-88%的产率得到产物2h-2w。值得注意的是,不饱和羰基化合物如烯基酮、丙烯醛类似物也可作为合适的偶联配偶体,以32-72%的产率得到相应的产物2x-2z, 2Aa-2Ac。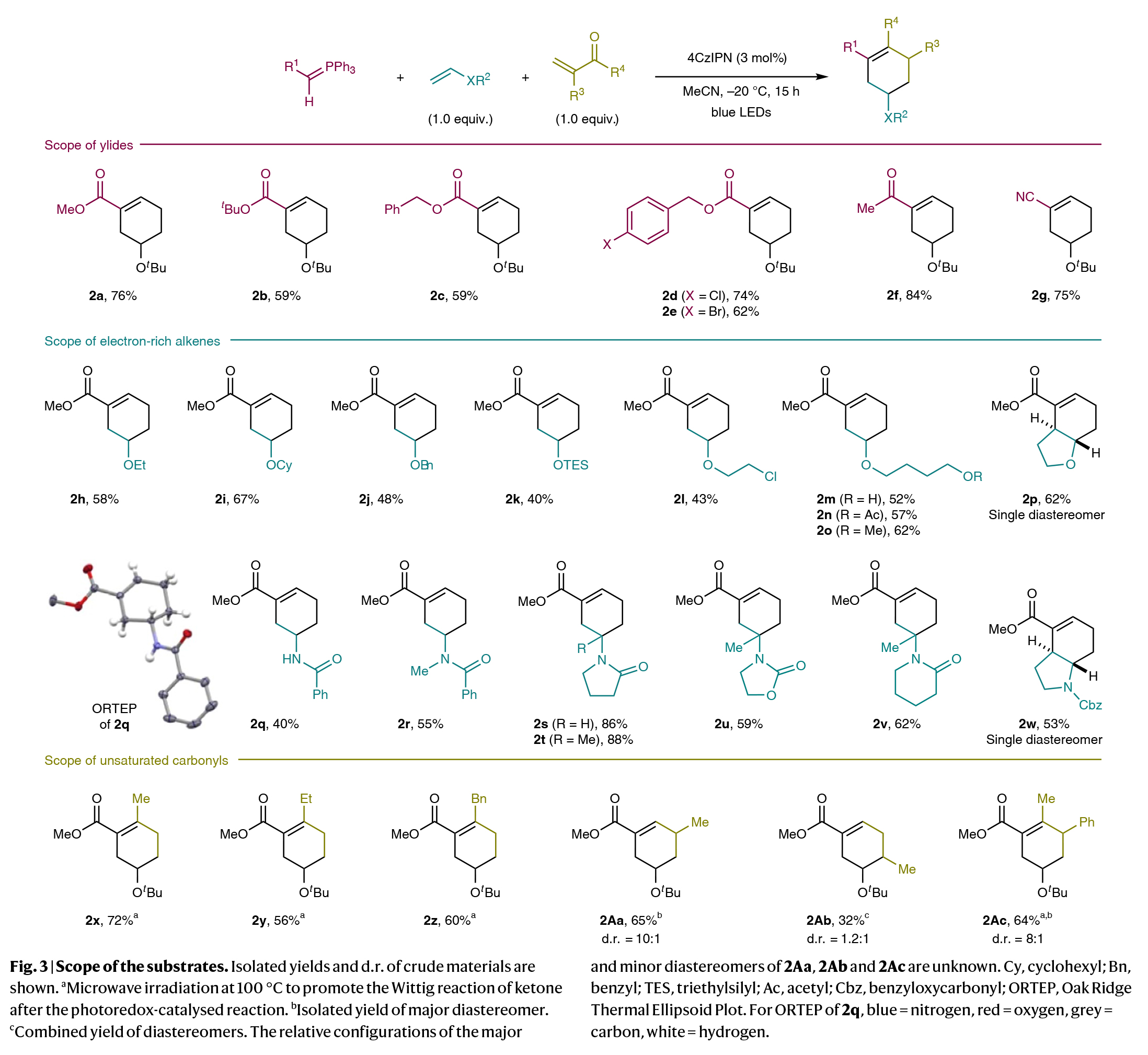
(图片来源:Nat. Synth.)
为了深入理解反应机理,作者进行了控制实验。方波伏安测量和Stern-Volmer淬灭实验表明,形式环加成过程是由激发态为4CzIPN与磷叶立德的单电子氧化触发的。为了从产生的自由基阳离子中更深入地了解反应途径,作者进行了DFT计算。如Fig. 4所示,RC (TS1-RC)与叔丁基乙烯基醚的加成过程很容易进行,而其通过去质子化生成叶立德自由基作为卡拜等价物(CE)似乎在能量上是不利的,且随后CE与叔丁基乙烯基醚的自由基加成则遇到了更高的能垒。此外,预估的pKa值表明,RC到CE的去质子化途径不太可能,因为RC的酸性远低于磷叶立德的共轭酸(1a·H)。随后丙烯醛与α-氧烷基自由基(Int1-CE和Int1-RC)加成得到的α-甲酰基烷基自由基(Int2-CE和Int2-RC)在热力学上表现出相似的能量过程。这两种途径通过单电子还原以及质子化或质子转移,得到官能团化的磷叶立德并经历了分子内Wittig反应。计算和实验研究表明,六元碳环的形成依赖于磷叶立德衍生的自由基阳离子的连续自由基加成和连续的极性关环过程。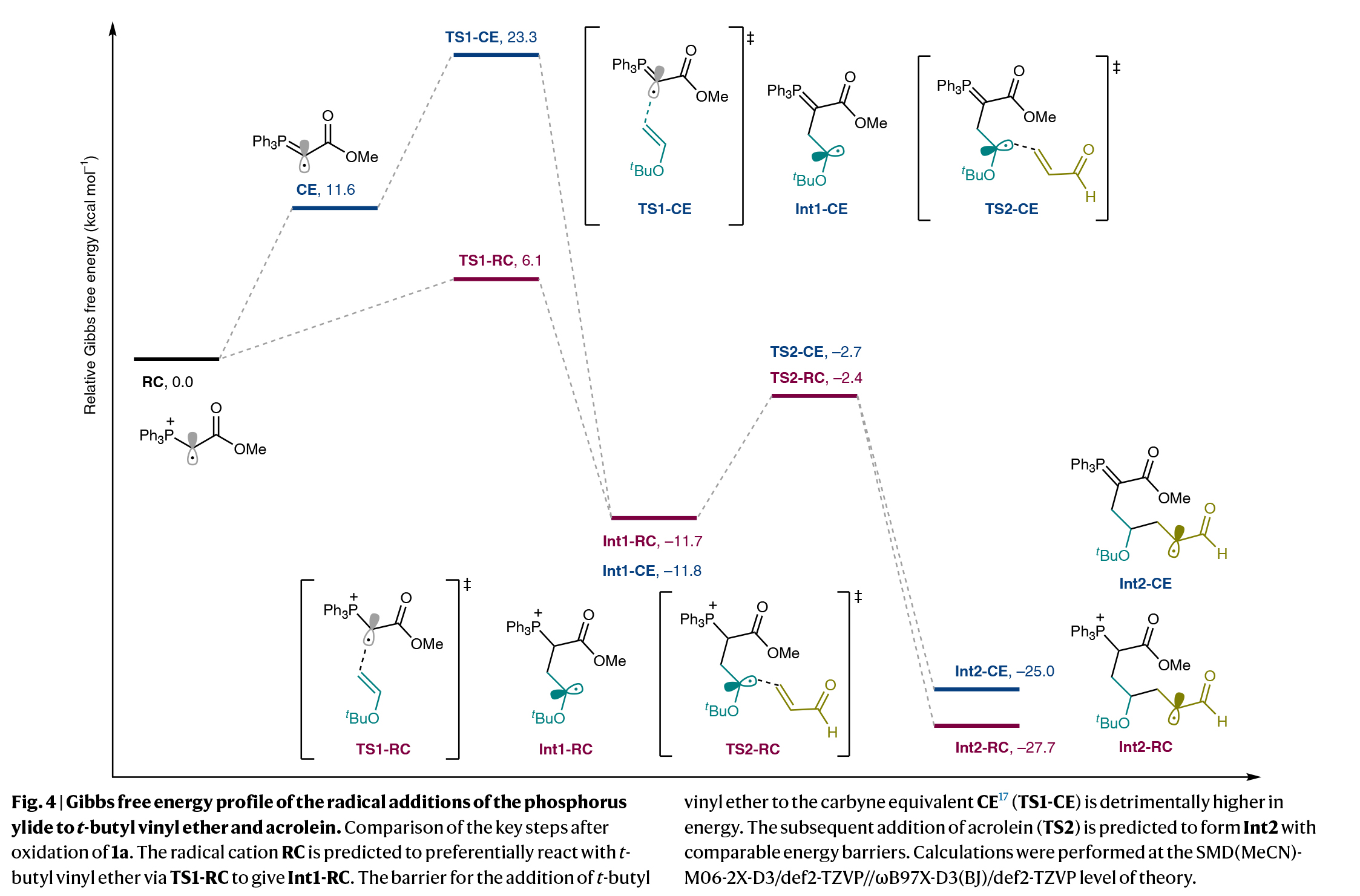
(图片来源:Nat. Synth.)
最后,作者进一步研究了一锅法四组分偶联反应的可能性(Fig. 5)。将磷叶立德1a、叔丁基乙烯基醚、丙烯酸甲酯以及催化量为4CzIPN的混合物置于乙腈中,在-20 oC的蓝光照射下组装成相应的双官能团化叶立德。随后加入多聚甲醛并在100 oC下进行微波照射,可以以72%的产率得到产物3a。尽管使用其他的醛,如苯甲醛作为第四组分反应时阻碍了其与中间体取代的叶立德化合物成键,但该策略可以适用于与不同的叶立德和烯烃偶联配偶体的偶联,说明了此转化在快速构建复杂分子骨架方面的显著特点,否则这些结构很难从现有的起始原料中获得。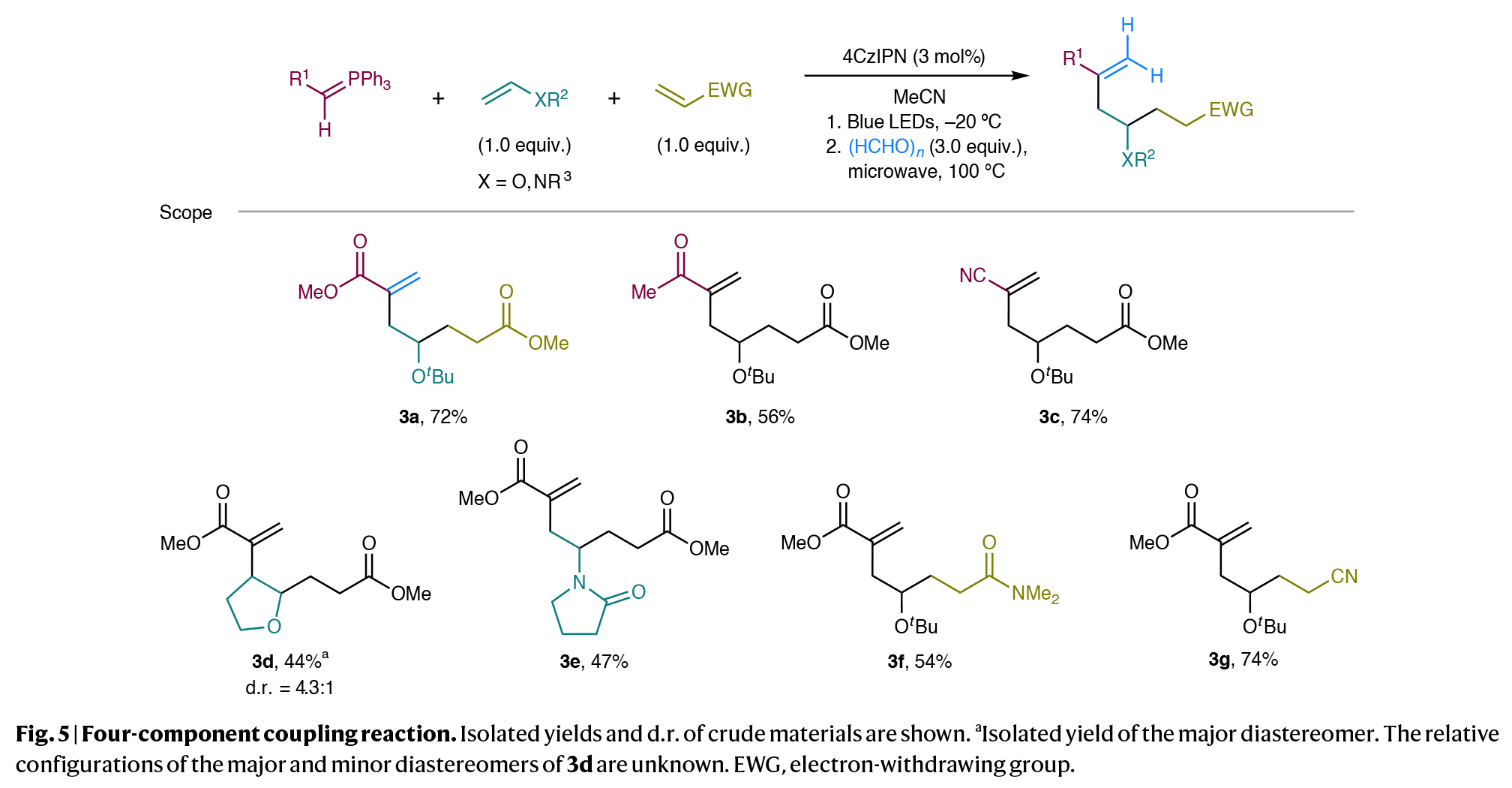
(图片来源:Nat. Synth.)
总结
Kohsuke Ohmatsu和Takashi Ooi课题组揭示了磷叶立德化合物利用光氧化还原极性反转构建三根化学键的潜在能力。磷叶立德在光催化下的卡拜反应性应用于多组分反应的发展,以与富电子烯烃和α,β-不饱和羰基化合物的形式环加成为代表,形成了不同官能团化的六元碳环。机理研究证实,在低温条件下,磷叶立德的单电子氧化以及由此产生的自由基阳离子的连续自由基加成途径,实现了自由基-极性交叉C-H烷基化和C=C成键环化。该方法为从简单易得的化合物高效合成复杂分子提供了一个新的策略。
文献详情:
Photocatalytic carbyne reactivity of phosphorus ylides for three-component formal cycloaddition reactions
Ryuhei Suzuki, Taiga Ando, Fritz Deufel, Kohsuke Ohmatsu*, Takashi Ooi*.
Nat. Synth., 2024
来源: 化学加
