
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 金属世界
金属世界 金属和合金材料在人类的文明发展史中具有举足轻重的地位,新型金属材料的出现是推动工业进步的重要因素。例如,由于纯银质地较软,古人通过在纯银中加入少量的铜可有效提高银的硬度,从而用于铸币;同样地,在铁中添加碳和铬等元素可以增强其强度和耐腐蚀性。在传统的合金设计理念中,一直沿袭在母体金属中加入相对少量的其他元素的基础合金化策略,但其揭示的合金种类只占整个多组分相图区域的一小部分,因此合金种类仍有较大的探索空间。
2004年,叶均蔚和Cantor课题组分别独立提出了一种全新的合金设计模式,开创了金属材料全新的研究领域——高熵合金[1]。这种设计模式打破了传统合金的设计思想,采用多种主要元素为基本组元。高熵合金被认为是最近几十年来合金化理论的三大突破之一。高熵合金通常包含5种及以上的主要元素,各主元的原子占比在5%~35%,其结构和性能在许多方面有别于传统的合金(即1~3种元素的合金)。得益于多组分带来的多元可调的原子结构,高熵合金许多特性不断地被发现,如卓越的超导性、超顺磁性、高稳定性和出色的储氢性能等。并且随着合成技术的发展,高熵合金的尺寸已经缩小到纳米级,这极大推动了高熵合金在催化领域的应用。由于多元素随机组合带来的巨大组成空间和复杂原子构型,相较于传统催化剂,高熵合金纳米催化剂具有多种吸附位点和近乎连续的结合能分布模型,可广泛应用于涉及多种中间步骤的复杂或串联反应[2]。
尽管高熵合金在催化领域具有巨大的潜力,但多元素排列组合造成了大量可能的原子构型,这对其原子结构的精准识别和实现以性能为导向的结构可控设计造成了阻碍。此外以熵为中心的讨论,特别是关于由不同元素组成决定的构型熵与催化性能的关系探索仍处于起步阶段。迄今为止,在对高熵合金纳米材料的微观结构、合成和表征等方面取得了一定进展。高熵合金的催化行为与其局域(表面等)结构高度相关。为了将对高熵合金纳米材料结构的理解与高效催化剂设计联系起来,本文结合目前高熵合金纳米材料的先进合成方法和表征技术,重点介绍对高熵合金纳米催化剂的基本见解,同时探讨高熵合金各组成元素和局域结构与催化反应之间的构效关系。
1、高熵合金的结构
传统合金通常由单一的基础金属和少数的其他元素组成,导致其具有可预测且有限的结构和性能。然而,高熵合金纳米材料通过将具有不同原子体积和价电子数的元素按特定比例组合在一起,会形成复杂的局域结构或晶体缺陷,例如晶格扭曲、位错、孪晶等特殊结构(图1)。
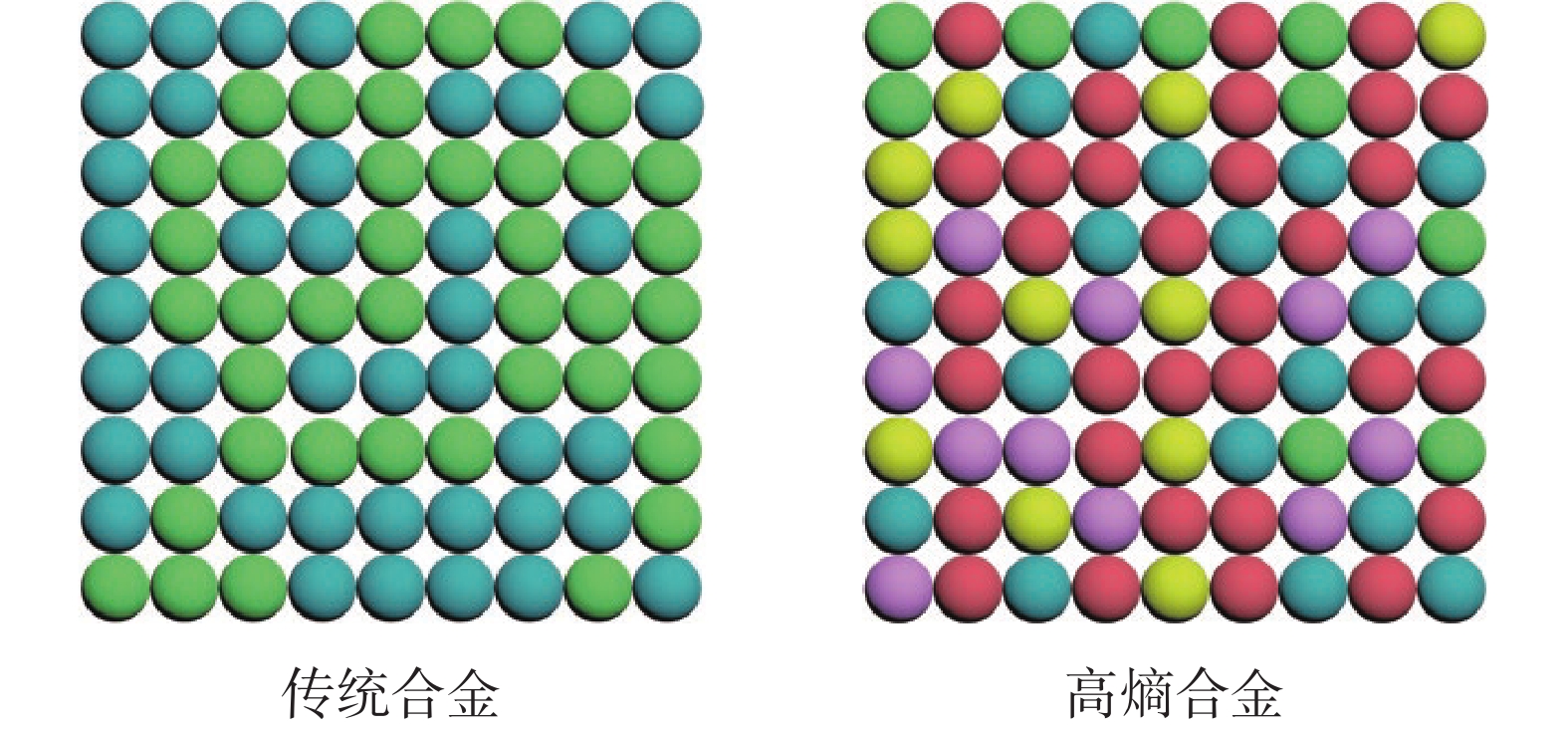
图1 传统合金和高熵合金的结构示意图
1.1 高熵合金的结构稳定性
根据近年来对高熵合金结构的研究,相比于其他的合金纳米材料,高熵合金具有更高的稳定性。高熵合金的高稳定性的原因可能主要来源于两个方面:(1)构型熵增加(高熵合金中元素的不同排列方式会导致较高的混乱度和熵增)导致的吉布斯自由能的降低;(2)迟滞扩散效应:缓慢的扩散速度使得高熵合金在高温时不易产生晶粒粗化及再结晶等不利影响,因而高熵合金具有较好的热稳定性。此外,原子在高熵合金中的扩散速度比在单一金属或传统合金中的扩散速度要慢得多,这主要是因为高熵合金的晶格畸变,提高了高熵合金在催化环境中的稳定性[3]。
从理论研究方面来说,高熵合金是具有高稳定性的,鉴于高熵合金在催化反应中可能面临的复杂工作环境,比如高温、高压以及氧化或还原环境等,从实验角度研究高熵合金稳定性和结构变化是至关重要的。Mori等[4]通过在真空中使用透射电子显微镜观察电子束辐照过程中高熵合金的稳定性。在连续拍摄的照片中分析原子位置的图像衬度,如图2所示,在边缘/角落位置,CoNiCuRuPd/TiO2的原子柱衬度变化相对较小,表明电子束并没有引起明显的结构改变(图2(a‒c))。相反,Pd/TiO2显示出由于辐照损伤引起的原子位移(图2(d‒f)),这表明高熵合金CoNiCuRuPd表面原子的稳定性明显优于单金属Pd。

图2 长时间辐照下高熵合金表面原子变化(a‒c)和钯颗粒表面原子变化(d‒f)[4]
同样的,如图3所示,Song等[5]通过原位透射电子显微镜研究了FeCoNiCuPt高熵合金纳米颗粒在400 °C下空气中的氧化过程。结果表明,高熵合金纳米颗粒的氧化速率明显低于单金属纳米颗粒。受柯肯达尔效应的影响,相比室温下的颗粒,在400 °C下,Fe、Co、Ni和Cu这些易氧化的过渡金属元素在表面富集,而贵金属Pt不易被氧化,富集在颗粒内部。

图3 高熵合金FeCoNiCuPt氧化过程原位电镜研究[5]
1.2 高熵合金的结构缺陷
高熵合金中存在多种结构缺陷,如空位、位错、晶界、层错等;也存在局部的元素偏析和有序结构。上述结构缺陷不仅会影响高熵合金的局部原子结构,也会破坏高熵合金中的局部化学环境。
在微观结构方面,高熵合金与传统合金相比的不同之处长期受到关注。如图4,相比于传统合金,由于高熵合金中多元素的复杂相互作用,如不同组元间原子尺寸、电负性以及混合焓的差异等可能引起局域应变、原子键合以及自由能的波动,最终在“看似元素分布无序的基体中”形成了局部的化学短程有序结构[6]。短程有序是指在材料中出现的局部结构有序性,这种有序性在原子级别上可能表现为特定元素的聚集、相互配对或在特定区域的排列。针对多组元合金的局域结构,提出更加宽泛的短程有序概念,即在几个原子尺度内,偏离无序原子排列的化学或拓扑结构组态。短程有序结构的存在可以影响高熵合金材料的力学性能、导电性、热导率等特性。通过调控短程有序,可以优化材料的性能,使其更适应特定的应用需求,如高温环境下的使用、耐腐蚀性能等。
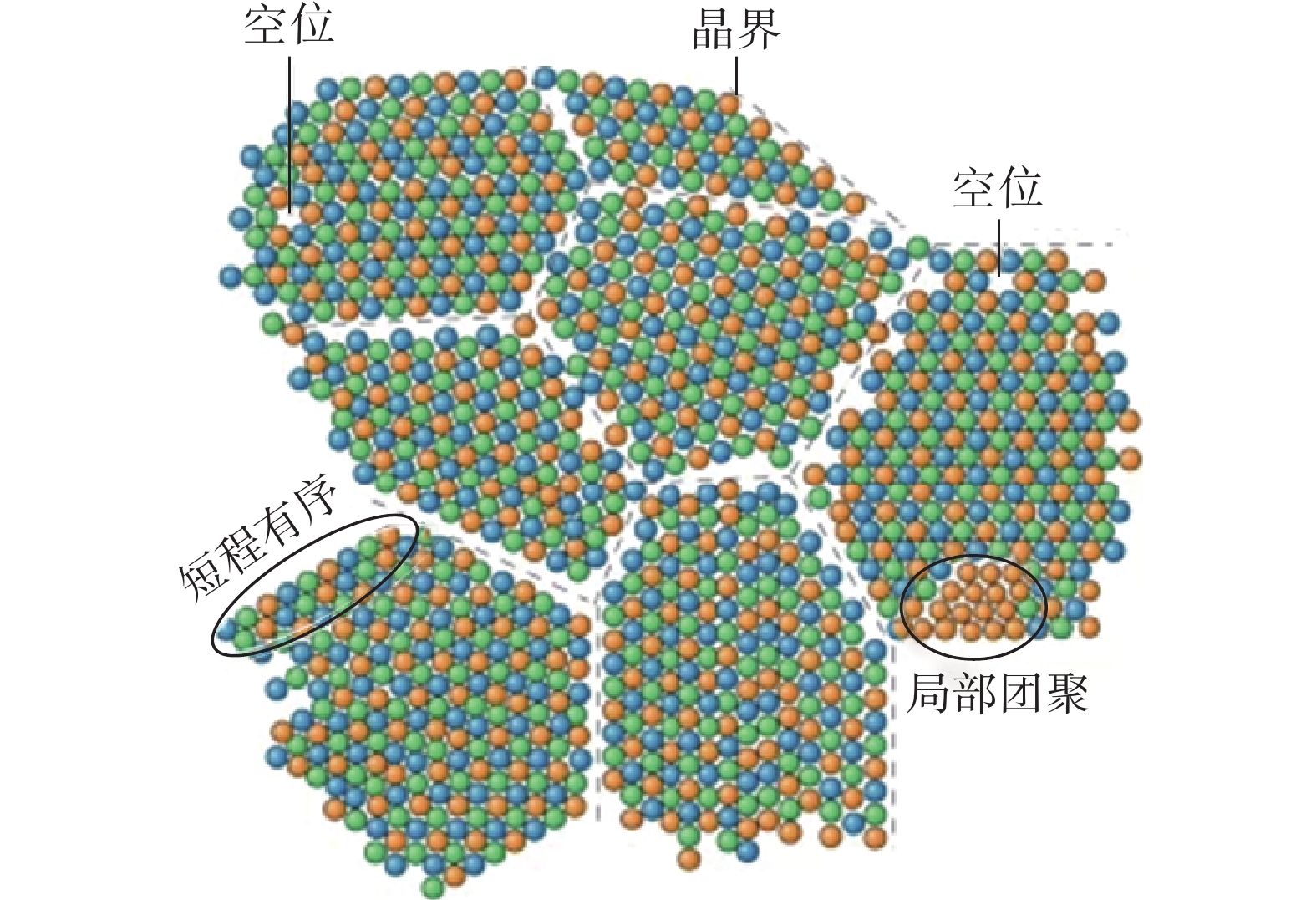
图4 高熵合金中的结构缺陷
2、合成方法
高熵合金纳米催化剂的各种特征,包括元素组成,粒径大小,纳米晶形貌以及相结构等,均受到合成策略和实验条件的影响。高熵合金催化剂的合成方法基本可以分为3类:(1)“冲击”型方法:通过快速冲击方法(快速升降温策略)等极端条件合成的纳米颗粒通常显示出单相结构;(2)湿化学方法:通过传统的湿化学方法制备的纳米颗粒大小均匀,形貌可控;(3)自上而下方法:与上述自下而上的方法相比,自上而下的方法是指通过将大块材料转化为纳米尺度颗粒或高表面积材料来合成高熵合金催化剂的方法。自上而下的方法一般更具可扩展性和简单性,但缺乏自下而上方法所示的精确控制。
2.1 “冲击”型方法
高温可以促进多个元素的均匀混合,然而在长时间冷却过程中仍存在相分离的可能,从而导致异质结构的形成。因此,将高温合成与快速淬火过程(即冲击过程)结合起来,确保各种元素均匀混合成单相高熵合金至关重要。高温碳热冲击法具有瞬态加热特征,是第1个可以高效合成均匀分散且具有单相结构高熵合金纳米颗粒的通用方法[7]。如图5所示,碳热冲击法包括以下步骤:(1)金属盐均匀分散在碳载体上;(2)金属前体经过高温环境加热分解,还原形成的金属在高温下均匀混合;(3)超快速淬火使高熵合金纳米颗粒均匀分散在碳载体上。通常情况下,碳热冲击法温度可以达到3000 K,持续时间短(毫秒级),冷却速率为105 K/s的数量级,但往往局限在碳载体上使用。
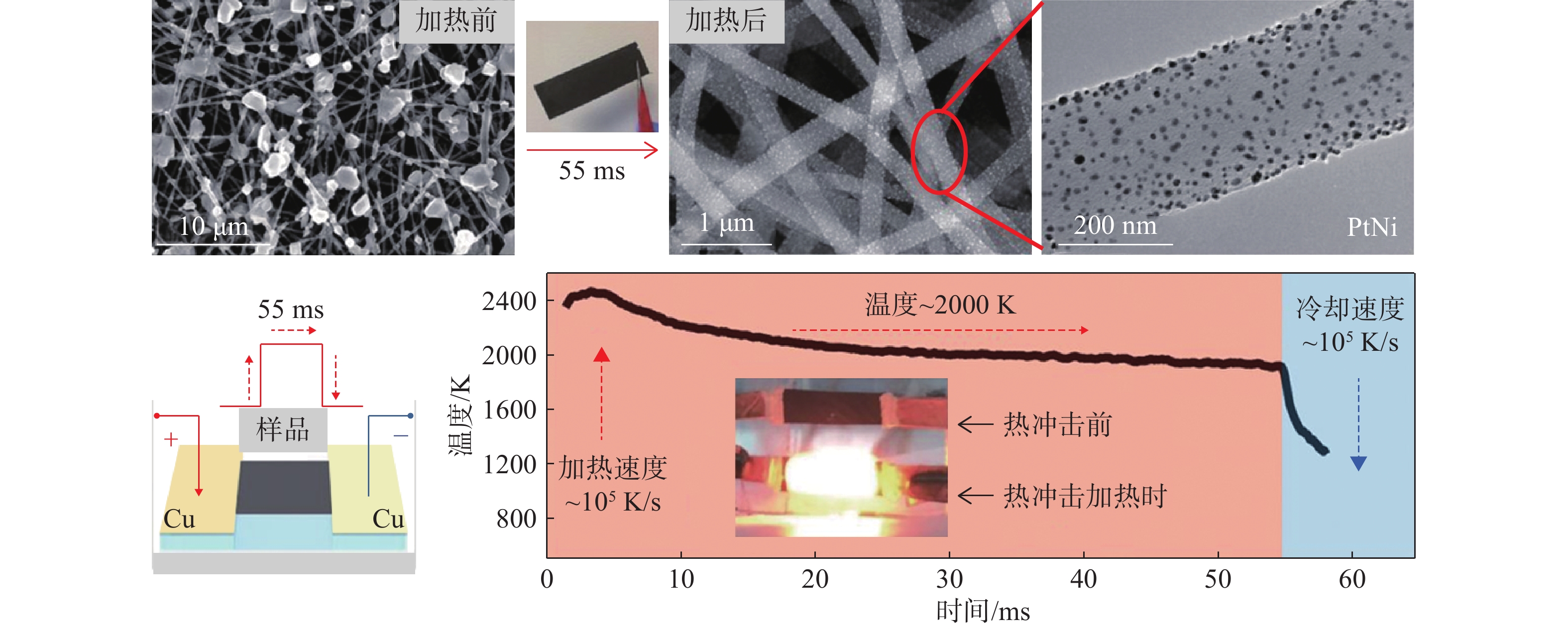
图5 碳热冲击法制备高熵合金纳米颗粒[7]
同时,通过设计元素组成和调节设备参数(如温度、退火时间、加热/冷却速率、碳基底物等),可以合成所需组成、大小和相结构的各种高熵合金催化剂。比如,碳载体表面缺陷的浓度控制着高熵合金纳米颗粒的大小和均匀性。冷却速率、保温时间可以调节形成纳米颗粒的相结构(异质结构或金属间化合物)。
除上述的碳热冲击法之外,微波加热法可以利用良好的导热系数和吸波能力部分还原氧化石墨烯,在几秒内通过微波加热可产生高达1850 K的平均温度,允许不同还原能力的金属同时热解还原,这种简便高效的方法为高熵合金纳米颗粒的合成提供了可行的路线[8]。同样的,像快速辐射加热,瞬态电合成以及等离子体和激光加热法这些具有明显的动力学驱动过程的快速冲击型合成方法也可以实现高效的高熵合金纳米材料的制造。
“冲击”型方法在高熵合金纳米颗粒的合成中具有很多优势:(1)实现快速制备,这种方法的制备过程通常比传统的熔融法更快,因为它不需要长时间的高温处理,可以在较短的时间内得到所需的高熵合金样品;(2)均匀性,冲击法可以通过合适的混合和处理步骤,实现相对均匀的元素分布,有助于提高合金的性能和稳定性;(3)高熵合金合成的多样性,这种方法可以用于制备不同类型的高熵合金,通过调整原始材料的组成和比例,可以制备出具有不同性质和应用的高熵合金;(4)适用性广泛,适用于各种金属体系,包括不同种类的金属元素,从而可以满足不同应用领域的需求。
2.2 湿化学合成法
湿化学合成是制备各种尺寸、形状、组成和结构明确的纳米晶体的最有效方法,相比于在特定气氛下的高温退火法,由于溶剂沸点的限制,湿化学合成通常在相对较低的温度下进行,这有助于避免高温下可能引发的颗粒聚集或烧结。如图6,对于混溶性良好的元素组合,通过湿化学方法可以相对精准的调控高熵合金纳米颗粒的尺寸、形貌以及暴露的晶面等,但在原子水平上对其尺寸和形态进行精确控制仍然是一个巨大的挑战[9]。
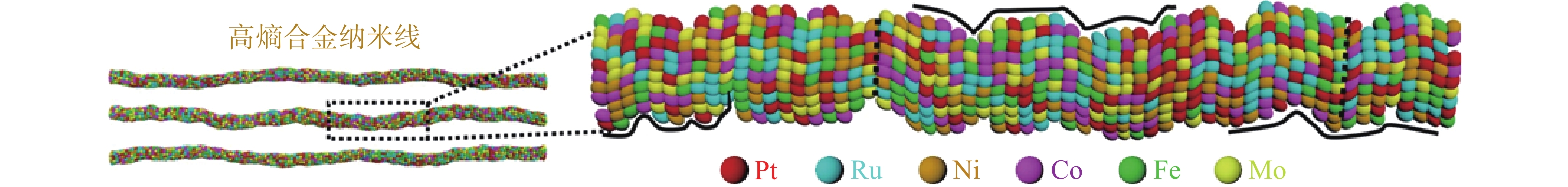
图6 湿化学法合成高熵合金纳米线[9]
当然湿化学法也存在很多缺点:(1)制备时间较长,与一些快速冲击型合成方法相比,湿化学合成法可能需要较长的反应时间,限制了其在快速制备领域的应用;(2)可能存在污染和杂质,在液相环境中,可能存在一些杂质或污染物,这可能会影响合金的纯度和性能;(3)可能需要后续处理,合成的高熵合金可能需要经过后续的热处理等步骤,以实现所需的相和性能。
2.3 自上而下的合成方法
磁控溅射是一种常用的薄膜制备技术,广泛应用于高熵合金的制备领域。通过选用不同的靶材,可以精准调控所制备高熵合金的化学成分。同时,通过调整溅射功率及其他工艺参数,还能够操控高熵合金薄膜的结晶状态。传统的直流磁控溅射可制备非晶态高熵合金薄膜,而高功率脉冲磁控溅射则有能力在薄膜生长过程中实现高熵合金的结晶态,这种方法对于快速制备高熵合金纳米颗粒具有独特优势[10]。
机械球磨法是一种无需或只需少量溶剂的工艺,通过固体之间的摩擦作用来制备纳米颗粒(图7)。相较于传统的湿法制备方法,这种工艺避免了大量使用金属盐和有机溶剂所带来的问题,减少了前体材料的浪费,还能降低中毒风险[11]。
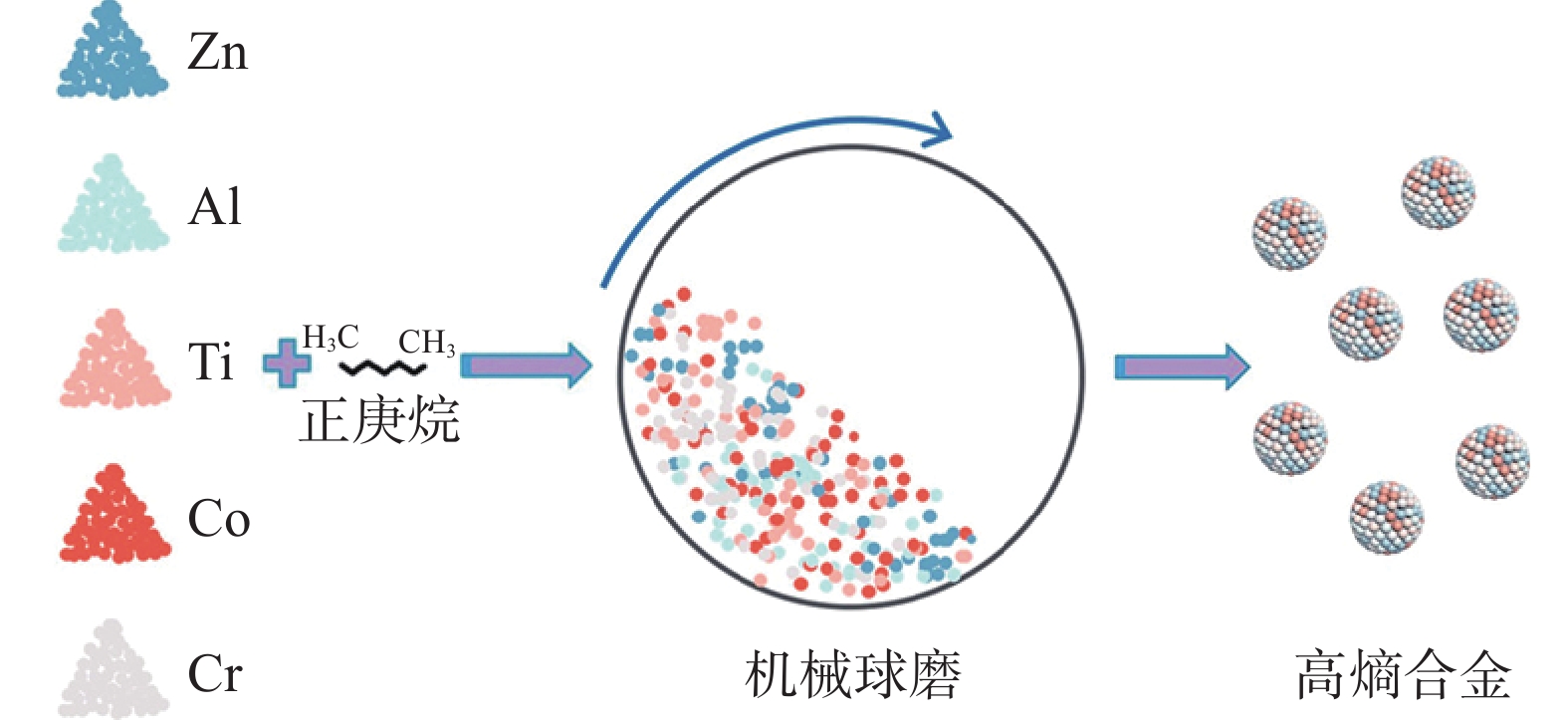
图7 机械球磨法合成高熵合金纳米颗粒
3、表征方法
高熵合金的性能与其结构密切相关。一方面,其巨大的构成空间为结构调整和设计带来巨大潜力;另一方面,复杂的多元素组成对其结构的准确理解提出了很大的挑战,因此迫切需要利用先进的表征方法来阐明高熵合金的原子和电子结构,主要表征方法见图8。
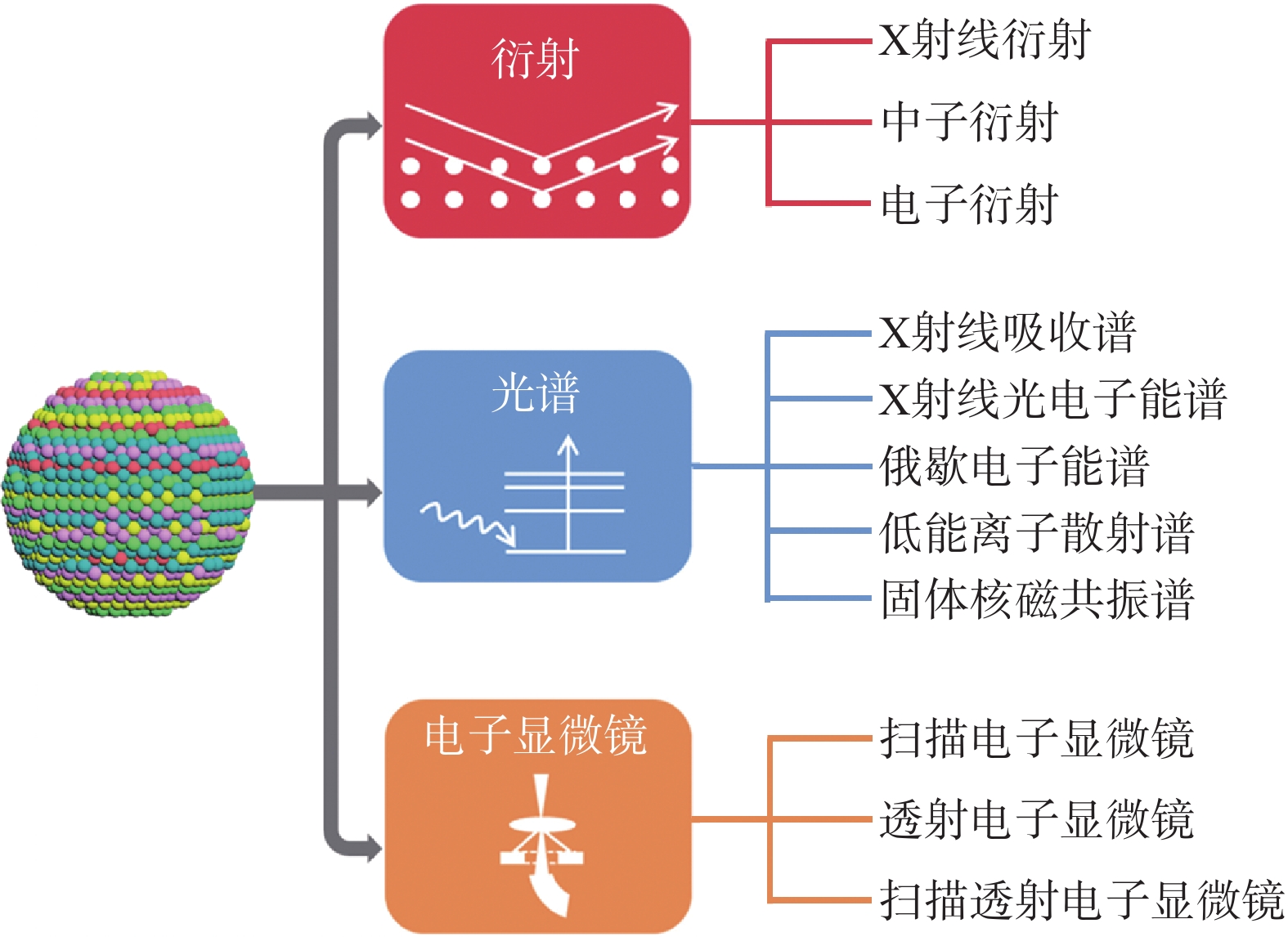
图8 高熵合金表征方法
3.1 衍射法
衍射技术被广泛应用于研究晶体材料的体相结构,其中X射线衍射(XRD)可以快速确定高熵合金晶体结构,以及在高熵合金中是否发生相分离。通过对X射线衍射谱图的精修,还可以确定各种结构参数,如晶格参数、结晶度、晶粒尺寸、优选取向和应变。然而,典型的实验室X射线衍射仪存在明显的局限性,在表征小尺寸纳米结构的高熵合金时,会出现衍射峰展宽的现象(图9),难以识别图中潜在的杂质峰[12]。为了克服这一缺点,采用高强度光源的同步加速器或基于中子衍射的技术会使展宽效应明显减弱,并以更高的精度提供有关高熵纳米颗粒中各种结构方面的原子级信息,包括畸变、原子位移、缺陷和晶格应变。
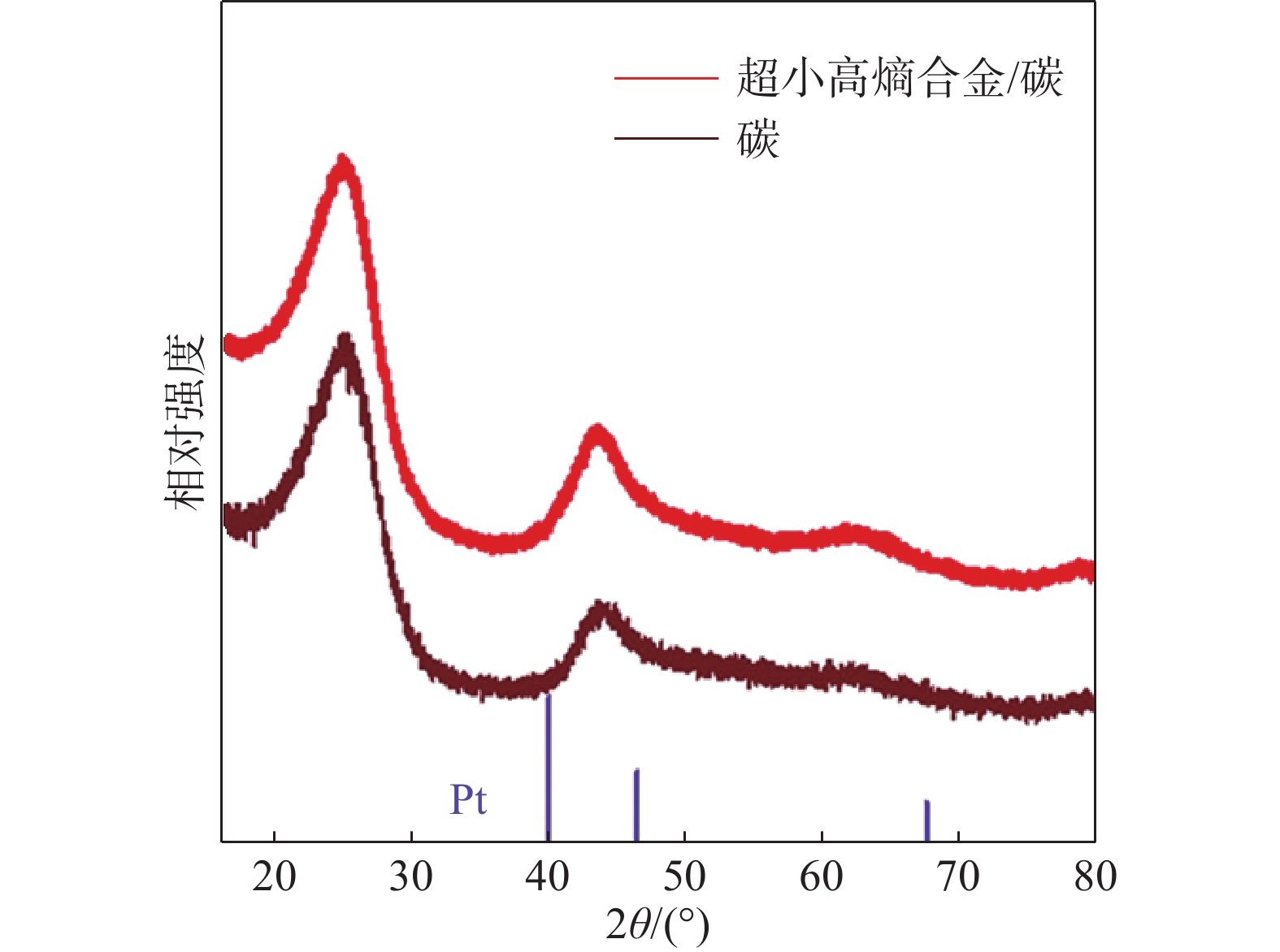
图9 高熵合金纳米颗粒X射线衍射图[12]
3.2 光谱法
涉及能级跃迁的光谱技术在揭示高熵合金的化学状态和电子结构方面非常有效。其中,X射线吸收光谱(XAS)是一种利用其X射线吸收特性对样品中的元素和化学成分进行批量测量的技术,包括X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)谱。这种测试方法对于解开高熵合金中特定吸收原子的电子轨道和配位环境至关重要,也可以揭示高熵纳米颗粒表面以及内部的电子结构,具有较好的灵敏度和更高的能量分辨率(图10)[12]。同时,通过它可以原位观察催化过程中高熵合金氧化态的变化,识别反应中间体的存在,并跟踪表面重建或中毒效应。除此之外,高熵合金表面和次表面上的元素组成及其氧化状态还可以通过X射线光电子能谱(XPS)测量。配备离子溅射技术后,XPS能够识别不同深度处高熵合金的化学组成和化学状态。其他光谱技术,如俄歇电子能谱(AES)和低能离子散射(LEIS),也可以提供有关高熵合金表面元素组成和表面电子结构的信息。
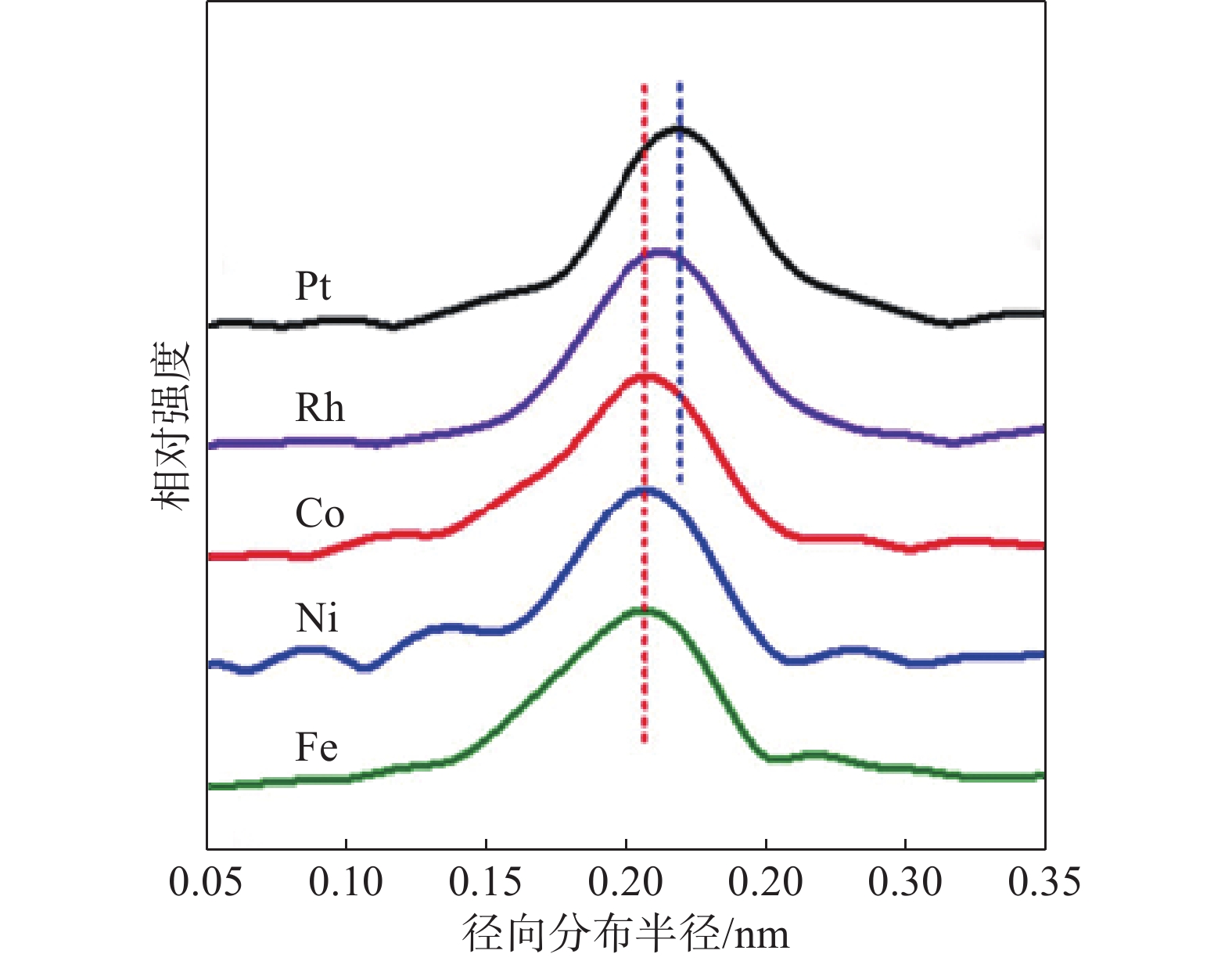
图10 高熵合金纳米颗粒扩展X射线吸收精细结构(EXAFS)谱[12]
3.3 电镜表征
尽管各种衍射和光谱表征方法可以提供高熵合金宏观尺度的信息,但获得单个纳米颗粒的结构和组成信息非常困难。然而,透射电子显微镜(TEM)能够以极高的空间分辨率,微电子伏特能量分辨率和毫秒时间尺度进行真实空间成像,最近已成为揭示原子结构和跟踪纳米材料动态演变的强大技术。不仅可以利用球差校正的扫描透射电子显微镜(STEM)实现原子分辨率的精准观测(图11)[12],还可以利用能量色散谱(EDS)探测局部区域的元素分布。

图11 原子分辨的高熵合金纳米颗粒STEM照片[12]
另外,其他基于TEM的高端电镜表征技术也应用到高熵合金结构的表征当中:四维扫描透射电子显微镜(4D-STEM)技术,它将STEM照片与扫描区域每个像素的电子衍射图相结合,从而能够快速表征高熵合金纳米颗粒的晶体学信息,如局部晶格畸变和表面应变等;此外电子能量损失光谱(EELS)也可以提供有关局部化学组成的信息,并且可以分析单个纳米颗粒表面和内部电子结构的差异。这些基于TEM的技术为表征高熵纳米颗粒的原子结构、局部组成、晶格畸变和动态行为提供了强大的工具,为了解其性质和优化其性能提供了关键信息。
4、高熵合金的催化应用
高熵合金纳米材料作为高活性、稳定的催化剂得到了广泛的研究。为了更好设计高熵合金催化剂,需要对其元素选择、元素相互作用和活性位点的确定进行更深入的研究。催化反应是一个涉及多个反应中间体和电子转移的多步骤过程。催化剂中许多相邻元素之间独特的相互作用是形成大量不同活性位点的基础,这些活性位点覆盖了广泛的吸附能范围,在高熵合金中不同活性位点的活性彼此非常接近,同时这也意味着可能只存在少量的高活性吸附位点,其他大量的活性位点具有较低的活性(图12(a)),优化其组成是实现吸附能可控调节的有效途径。
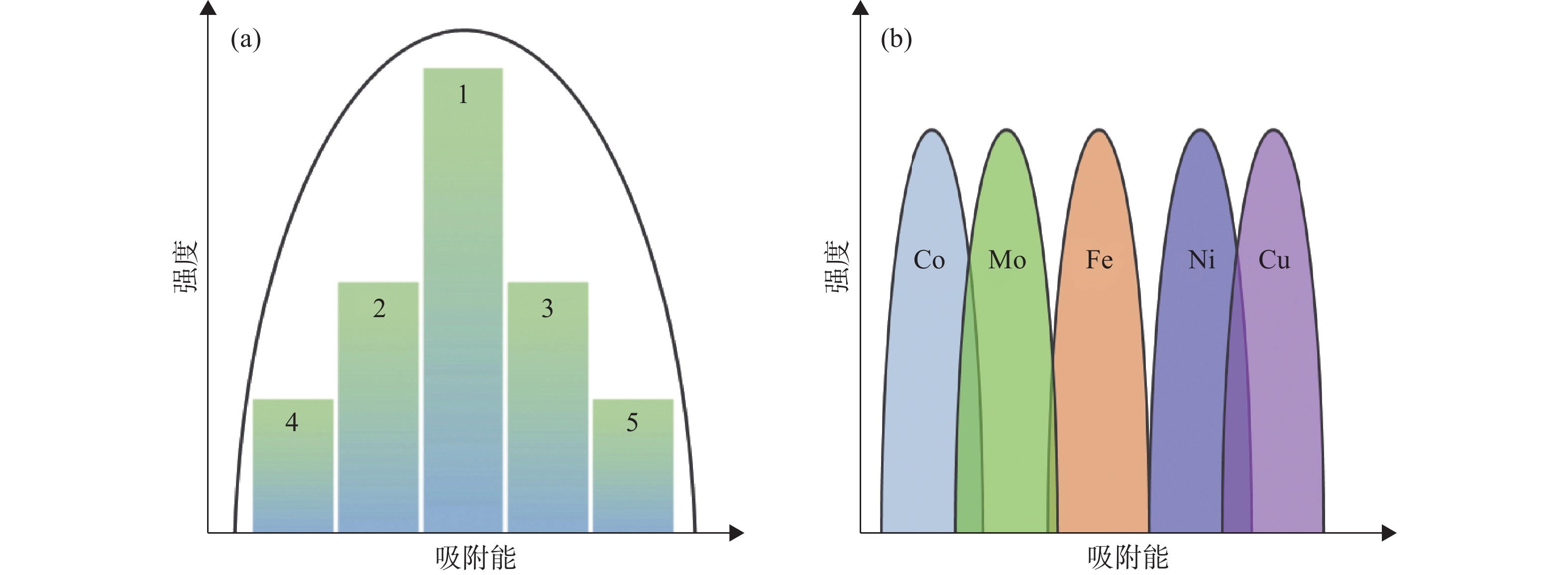
图12 (a)高熵合金活性位点的分布示意图,强度取决于类似活性位点的数量;(b)取代或增加高熵合金元素对吸附能分布模型的影响
如图12(b),高熵合金本征的多元素组成空间使其具有独特的微调吸附能的能力,通过调整元素组成可以显著调整吸附能分布曲线,单个元素(Co、Mo、Fe、Ni和Cu)的结构和吸附位点相对固定,因此它们的吸附能分布图通常会呈现尖锐的峰值。然而,当多种元素混合成高熵合金(CoMoFeNiCu)时,它们的吸附能会通过电子杂化转变成拓宽的、多峰的、近乎连续的形式。在纳米颗粒表面存在着由多个元素组成的具有不同性质的活性位点,在催化反应中,反应物在某一类特定反应位点完成第一步反应后,其反应中间体可以在其他反应位点进行后续反应(图13),通过合理选择元素构型和组成,可是实现特定反应选择性和活性的明显提升。简而言之,多数催化反应都经历了对反应物的吸附、反应物的解离(或转化)、不同中间体的转化以及产物的形成和解吸过程,那么在高熵合金中,可以产生不同的活性位点来分别加速上述过程,最终大大提高催化反应的速率,并且其催化性能高于单个元素的叠加[13]。
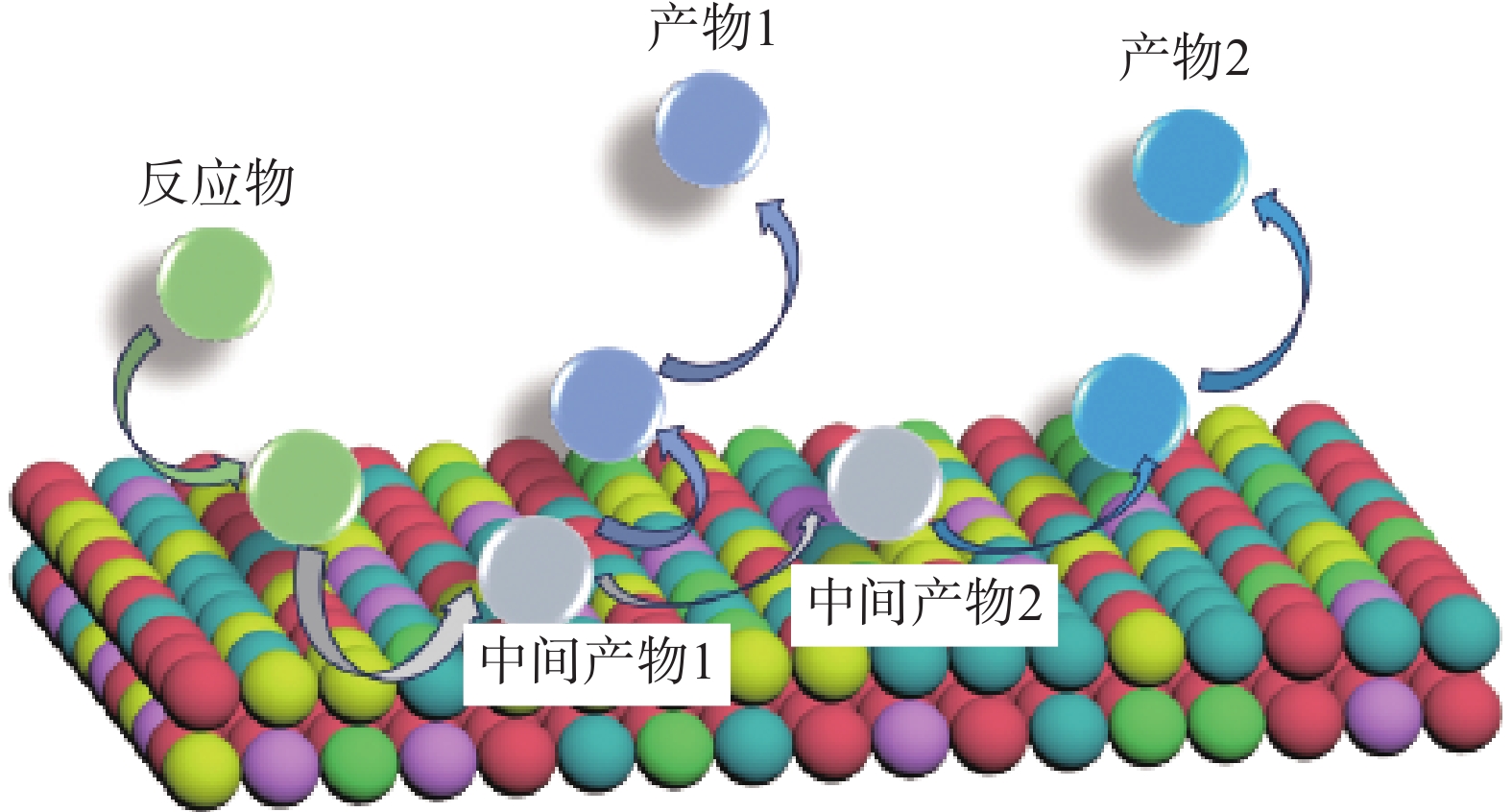
图13 高熵合金化剂表面催还反应过程示意图
例如,催化氨气分解为氢气和氮气这一反应无论是在能源转化利用,还是氨废弃处理等方面都有重要的意义。贵金属钌仍是目前氨分解反应的主流催化剂,但其高成本和稀缺性严重制约了应用规模。钴钼(CoMo)合金纳米晶合成成本低,表面有丰富多样的混合吸附位点,是氨气分解的候选材料。但是受限于其二元相图,仅能合成异质结构和组分确定的金属间化合物,致使CoMo合金的组成比例无法可控调节,无法达到最佳的催化效果。那么,利用高熵合金的本征特性,就可以可控合成Co/Mo比例可调的五元CoMoFeNiCu高熵合金纳米颗粒,达到最佳的催化效果。如图14所示,在机理研究方面,Co元素组成比例高的高熵合金催化剂结合氮物种能力过弱,无法达到氢气脱附的动力学壁垒,而Mo元素组成比例高的催化剂结合氮物种的能力过强,无法实现氮气的重组和脱附。但特定Co/Mo比例的催化剂(HEA-Co25Mo45)可实现氮的结合能为79 kJ/mol,非常接近于钌纳米晶体(84 kJ/mol),每克HEA-Co25Mo45的氨气分解速率为22.1 g/h,相较Co–Mo二元催化剂提高了约19倍。值得注意的是,CoMoFeNiCu高熵合金纳米颗粒在不同温度下(573~1000 K)也显示出极好的催化稳定性[14]。
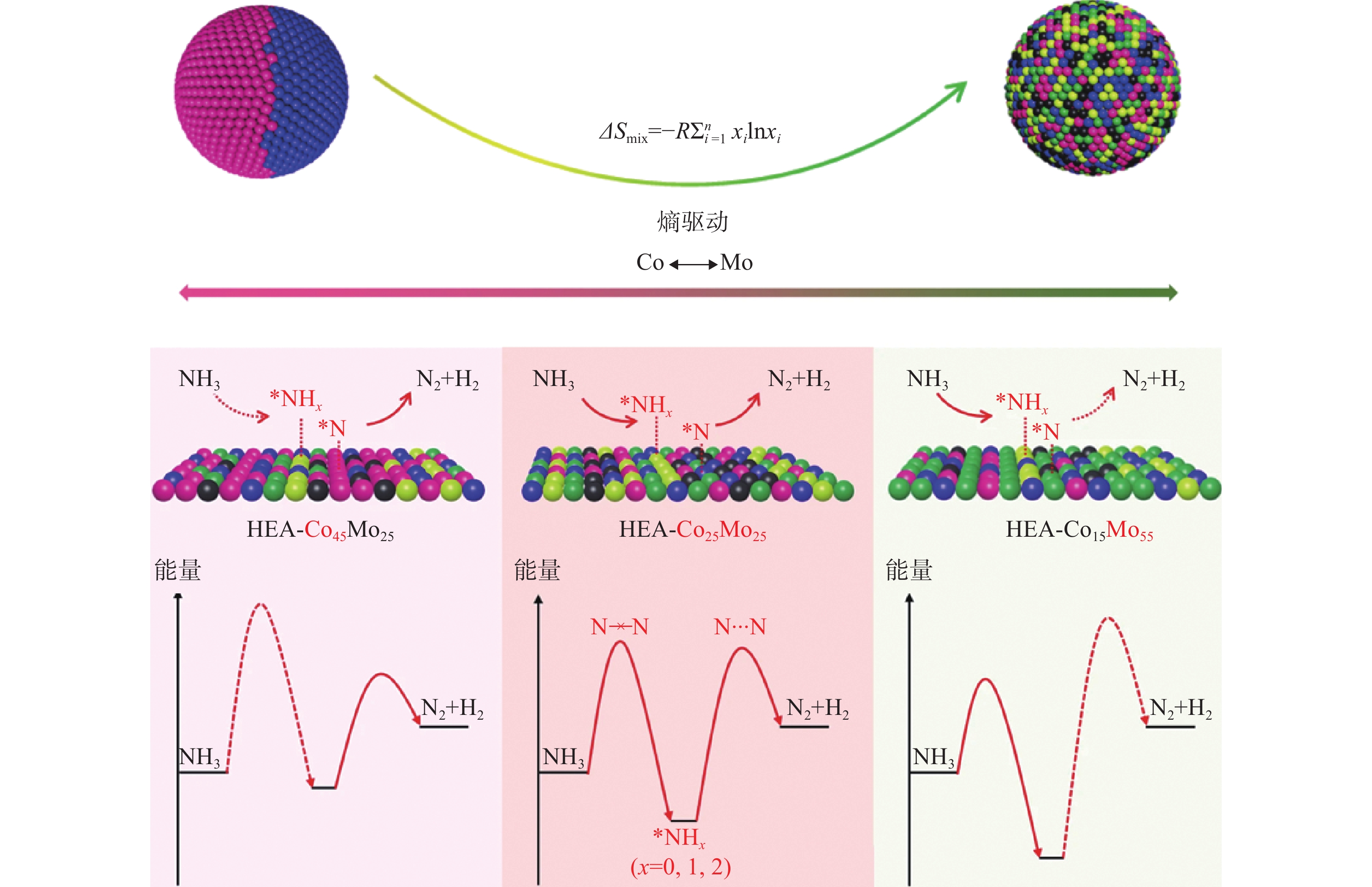
图14 氨气在不同高熵合金表面上的吸附脱附能比较[14]
5、总结与展望
高熵合金纳米催化剂是一类在催化反应中具有潜在应用的材料。然而,目前仍然存在一些不足之处,同时也有许多未来的发展前景。
5.1 目前存在的不足
(1)设计复杂性:高熵合金由多种不同元素组成,其合金设计和制备相对复杂,无法准确的查明每个原子的精确位置、配位环境和局部结构。选择合适的元素、比例和配位方式对于获得优越的催化性能至关重要,但这需要大量的试验积累和计算模型优化。
(2)活性和选择性:虽然高熵合金纳米催化剂在某些催化反应中表现出色,但并不一定在所有反应中都具有优越的活性和选择性。需要更深入的研究来了解其催化机理和反应途径。
(3)稳定性和耐腐蚀性:高熵合金在一些催化环境中可能面临腐蚀和热稳定性的问题。不同元素的相互作用可能导致催化剂的稳定性降低,限制其长期使用。
(4)制备技术:目前,制备高熵合金纳米催化剂的制备方法仍在不断发展中。不同的合金制备方法可能会影响催化性能,因此需要进一步研究最佳的制备方法。
5.2 未来的展望
(1)精准设计:随着材料计算和机器学习的发展,可以更精确地预测和设计高熵合金纳米催化剂的性能。这将有助于减少试验成本和时间,提高合金设计的成功率。
(2)催化机理研究:进一步研究高熵合金纳米催化剂的催化机理,可以帮助优化其结构和成分,提高活性和选择性。
(3)表征技术的发展:高熵合金纳米催化剂的性能与其微观结构密切相关。发展更先进的表征技术,如原位观察、原子分辨率显微镜等,有助于深入理解其结构–性能关系。
(4)可持续性和环保性:未来的发展还应考虑高熵合金纳米催化剂的可持续性和环保性。研究如何降低催化剂中的稀有和有毒元素含量,以及如何实现废弃物的可回收利用,将是重要的方向。
(5)多功能性催化剂:高熵合金纳米催化剂可能在不同反应中发挥多种功能。未来的研究可以探索如何设计具有多功能性的高熵合金纳米催化剂,从而在多个催化应用中得到应用。
虽然高熵合金纳米催化剂还存在一些挑战和不足,但随着材料科学和催化化学的不断发展,我们可以期待在设计、性能优化和应用方面取得更大的突破。
参考文献
[1] Yeh J W, Chen S K, Lin S J, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater, 2004, 6(5): 299 DOI: 10.1002/adem.200300567
[2] Yao Y G, Dong Q, Brozena A, et al. High-entropy nanoparticles: synthesis-structure-property relationships and data-driven discovery. Science, 2022, 376(6589): 151
[3] George E P, Raabe D, Ritchie R O. High-entropy alloys. Nat Rev Mater, 2019, 4(8): 515 DOI: 10.1038/s41578-019-0121-4
[4] Mori K, Hashimoto N, Kamiuchi N, et al. Hydrogen spillover-driven synthesis of high-entropy alloy nanoparticles as a robust catalyst for CO2 hydrogenation. Nat Commun, 2021, 12(1): 3884 DOI: 10.1038/s41467-021-24228-z
[5] Song B A, Yang Y, Rabbani M, et al. In Situ oxidation studies of high-entropy alloy nanoparticles. ACS Nano, 2020, 14(11): 15131 DOI: 10.1021/acsnano.0c05250
[6] Chen X F, Wang Q, Cheng Z Y, et al. Direct observation of chemical short-range order in a medium-entropy alloy. Nature, 2021, 592(7856): 712 DOI: 10.1038/s41586-021-03428-z
[7] Yao Y G, Huang Z N, Xie P F, et al. Carbothermal shock synthesis of high-entropy-alloy nanoparticles. Science, 2018, 359(6383): 1489 DOI: 10.1126/science.aan5412
[8] Qiao H Y, Saray M T, Wang X Z, et al. Scalable synthesis of high entropy alloy nanoparticles by microwave heating. ACS Nano, 2021, 15(9): 14928 DOI: 10.1021/acsnano.1c05113
[9] Zhan C H, Xu Y, Bu L Z, et al. Subnanometer high-entropy alloy nanowires enable remarkable hydrogen oxidation catalysis. Nat Commun, 2021, 12(1): 6261 DOI: 10.1038/s41467-021-26425-2
[10] Löffler T, Meyer H, Savan A, et al. Discovery of a multinary noble metal-free oxygen reduction catalyst. Adv Eng Mater, 2018, 8(34): Art No. 1802269
[11] Nellaiappan S, Katiyar N K, Kumar R, et al. High-entropy alloys as catalysts for the CO2 and CO reduction reactions: experimental realization. ACS Catal, 2020, 10(6): 3658 DOI: 10.1021/acscatal.9b04302
[12] Feng G, Ning F H, Song J, et al. Sub-2 nm ultrasmall high-entropy alloy nanoparticles for extremely superior electrocatalytic hydrogen evolution. J Am Chem Soc, 2021, 143(41): 17117 DOI: 10.1021/jacs.1c07643
[13] Li H D, Lai J P, Li Z J, et al. Multi-sites electrocatalysis in high-entropy alloys. Adv Funct Mater, 2021, 31(47): Art No. 2106715
[14] Xie P F, Yao Y G, Huang Z N, et al. Highly efficient decomposition of ammonia using high-entropy alloy catalysts. Nat Commun, 2019, 10(1): 4011 DOI: 10.1038/s41467-019-11848-9
基金项目:国家重点研发计划资助项目(2021YFA1202300);国家自然科学基金资助项目(52371223,52101255);武汉市知识创新专项。
作者简介:姚永刚(1987—),华中科技大学教授,华中学者,国家四青人才。长期从事瞬态高温合成与制造技术,特别是新型能源材料的设计开发与低碳快速制造,助力国家能源转型及碳中和战略。成果在《Science》(封面 )、《 Science》(综 述)、《Nature》、《NatureNanotechnology》、《Nature Catalysis》等期刊发表,论文总被引用10000余次,入选斯坦福大学“全球前2%顶尖科学家”及科睿唯安“高被引科学家”榜单,并获得美国“2020 R&D 100 award”,2022 Metals Young InvestigatorAward,2022 届阿里达摩院“青橙奖”(化学材料类)及《麻省理工科技评论》“35岁以下科技创新35人”中国区先锋者称号。通信地址:华中科技大学材料科学与工程学院,430074。E-mail:yaoyg@hust.edu.cn。
来源: 金属世界
