
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
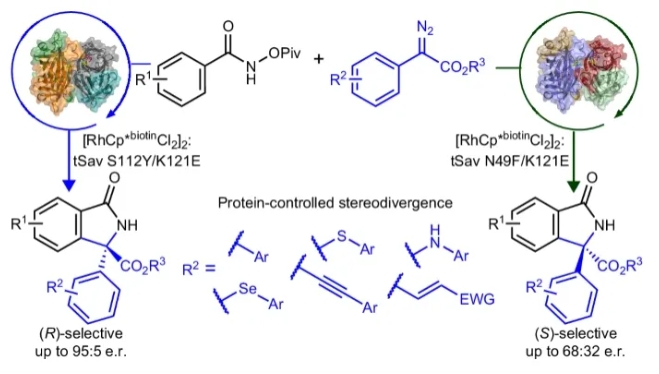
近日,印度理工学院孟买校区(Indian Institute of Technology Bombay)Prasenjit Bhaumik课题组和Debabrata Maiti课题组设计和发展了一个链霉亲和素-生物素-铑(III)催化体系,使用N-(特戊酰氧基)苯甲酰胺和芳香重氮酯为起始原料,以高达95:5的对映体比例实现了手性异吲哚酮的合成。此混合催化体系可以兼容多种不同的重氮酯,实现了具有多种有用官能团(联苯、硫醚、硒醚、胺、烯烃和炔烃)的手性异吲哚酮的合成。机理研究表明该转化涉及一个导向的内球C-H活化过程以及随后的重氮插入过程。此外,作者通过对链霉亲和素与Rh(III)辅因子的高分辨晶体结构进行分析设计出了N49位点的突变体,从而实现了对映发散合成。相关成果发表在Nat. Synth.上,文章链接DOI:10.1038/s44160-024-00533-5。
(图片来源:Nat. Synth.)
正文
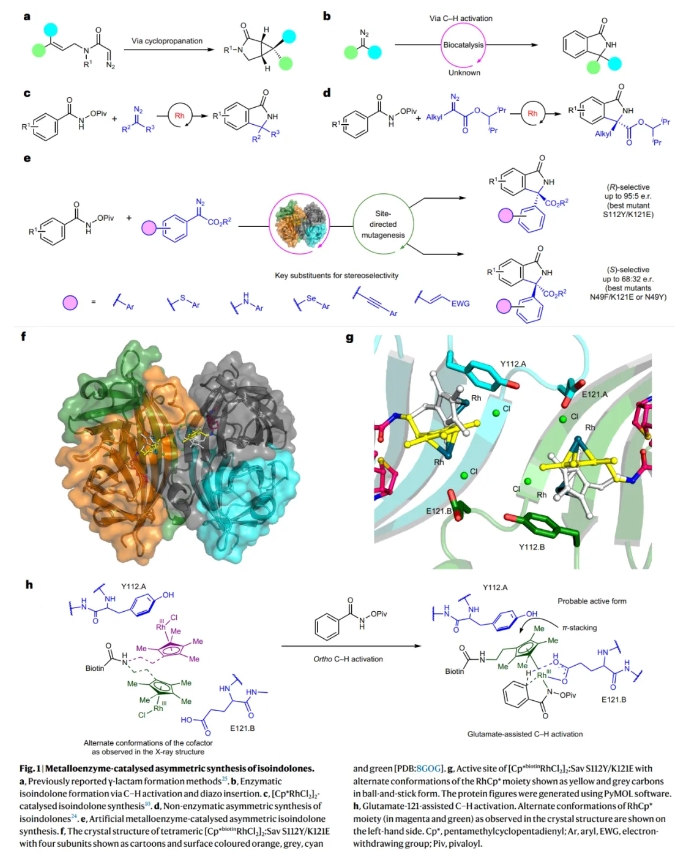
将酶催化和过渡金属催化结合在人工金属酶内可以有效拓展自然界中的未知化学转化。其可以通过在蛋白质活性位点内对反应催化中心进行识别,从而通过蛋白质工程在实现协同催化的同时得到良好的选择性,因此有效解决了不对称有机金属催化中的几个关键问题。链霉亲和素是一种同源四聚体蛋白质,其可以与生物素化的金属络合物结合在一起作为人工金属酶应用在多种非天然转化中。然而到目前为止使用基于链霉亲和素的人工金属酶来对映选择性的合成异吲哚酮仍未实现。最近,印度理工学院孟买校区Prasenjit Bhaumik课题组和Debabrata Maiti课题组发展了链霉亲和素-生物素-铑(III)催化体系,使用N-(特戊酰氧基)苯甲酰胺和芳香重氮酯为起始原料,以高达95:5的对映体比例实现了手性异吲哚酮的合成(Fig. 1)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
(图片来源:Nat. Synth.)
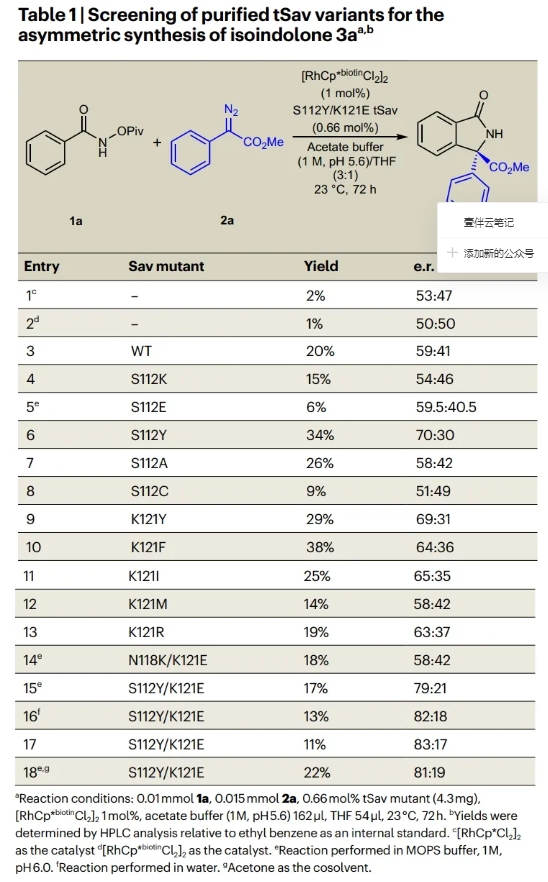
首先,作者选择N-(特戊酰氧基)苯甲酰胺1a和芳香重氮酯2a作为模板底物对反应条件进行了探索(Table 1)。通过对一系列酶突变体进行筛选,作者发现当使用1a (0.01 mmol), 2a (0.015 mmol), Sav S112Y/K121E mutant (0.66 mol%, 4.3 mg), [RhCp*biotinCl2]2 (1 mol%), MOPS缓冲液(3-(N-morpholino)propanesulfonic acid) (1 M, pH 6.0) 162 μl, 丙酮 54 μl, 23 °C反应72小时可以以22%的产率,81:19 e.r.得到手性异吲哚酮3a。突变体S112Y/K121E中酪氨酸和谷氨酸的协同作用对于反应的对映选择性至关重要。值得注意的是,单独使用[RhCp*Cl2]2和[RhCp*biotinCl2]2在醋酸盐缓冲溶液中催化是无法形成3a的。
(图片来源:Nat. Synth.)
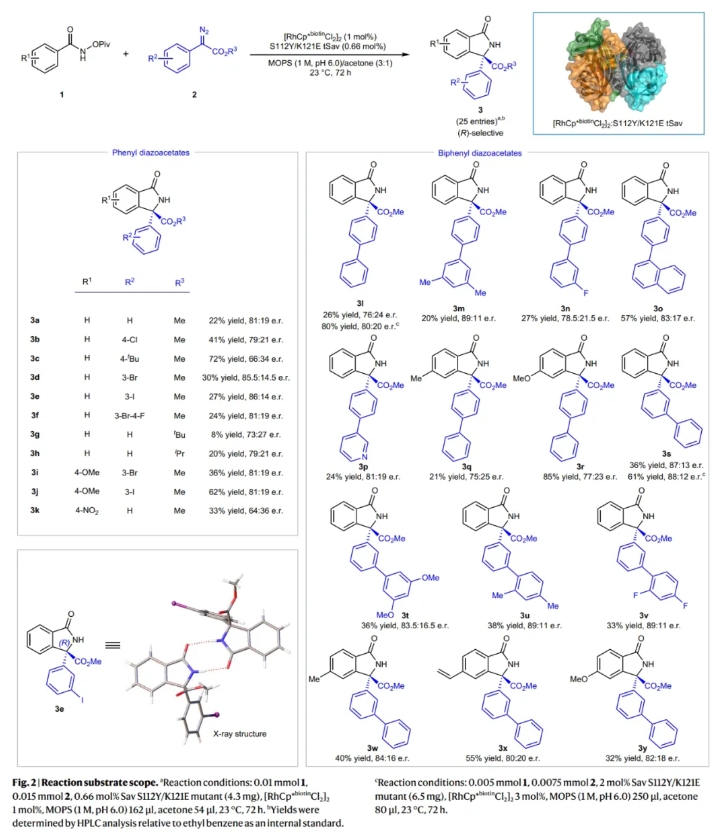
在得到了最优反应条件后,作者对此转化的底物范围进行了考察(Fig. 2)。实验结果表明,不同取代的N-(特戊酰氧基)苯甲酰胺和芳香重氮酯均具有良好的兼容性,以8-72%的产率,良好的对映选择性得到相应的手性异吲哚酮产物3a-3y。其中包括硝基、卤素、烷氧基和烷基等不同基团均可兼容。此外,作者通过X射线单晶衍射和HPLC分离证实了产物3e的绝对构型为R构型,这主要是由于S112Y/K121E的使用。通常来讲,苯甲酰胺的对位连有给电子基团(甲基,甲氧基等)时选择性相对较低。此外,在芳基重氮酯的对位连有Cl和tBu时产率相对较高。
(图片来源:Nat. Synth.)
接下来,作者对一系列结构复杂的芳基重氮酯的兼容性进行了考察(Fig. 3)。实验结果表明,包括3号位含有芳基硫、芳基胺、芳基硒和芳基乙炔以及4号位含有甲基、甲氧基、苯基、乙烯基以及硝基等取代的芳基重氮酯均可顺利实现转化,以合成有用的产率(8-67%)和中等的对映选择性得到相应的产物3aa-3bd。此外,将催化剂的量提升至3 mol%时,产物的产率可以增加两倍以上,而对映选择性几乎保持不变。
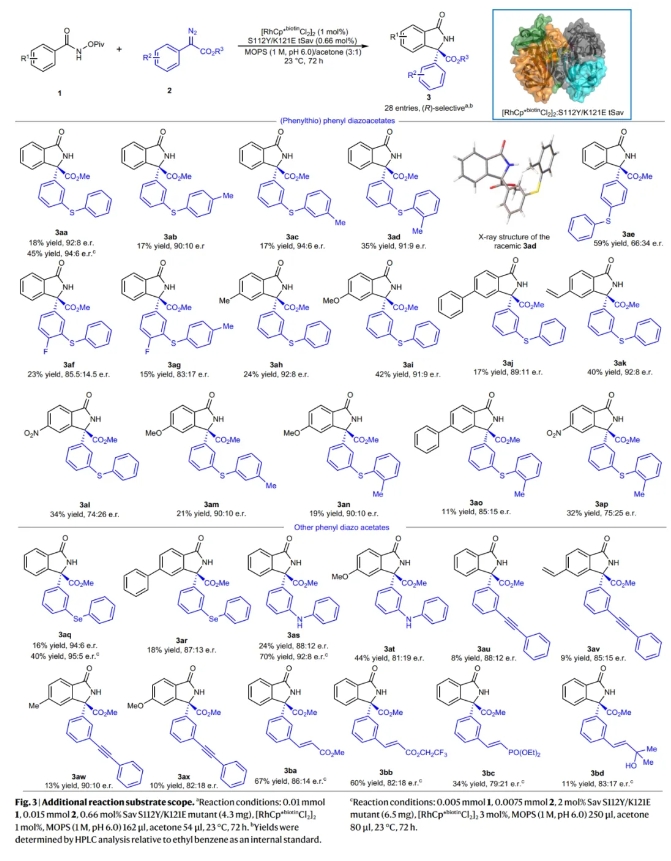
(图片来源:Nat. Synth.)
由于作者从[RhCp*biotinCl2]2:Sav S112Y/K121E络合物的高分辨晶体结构中观察到残基45-52非常接近反应中心,并在反应过程中大概率可以影响底物的靠进,从而影响反应的对映选择性结果。特别是距离辅因子5 Å(NCB到Cp*)的N49残基。因此作者对WT和S112Y/K121E骨架的N49残基进行突变得到了六个单突变体、五个三突变体和一个双突变体。其中,单突变体 N49V、N49I和N49Y和双突变体N49F/K121E与S112Y/K121E相比逆转了对映选择性,得到(S)-3aa为主要的立体异构体。此外,对于3s和3as来讲,N49Y比N49F/K121E更有效。而在所有其它情况下,突变体N49F/K121E倾向于相反的立体选择性(3a、3aa-3aq和3au)。上述结果均表明N49残基可以决定与重氮酯反应的面选择性(Fig. 4)。
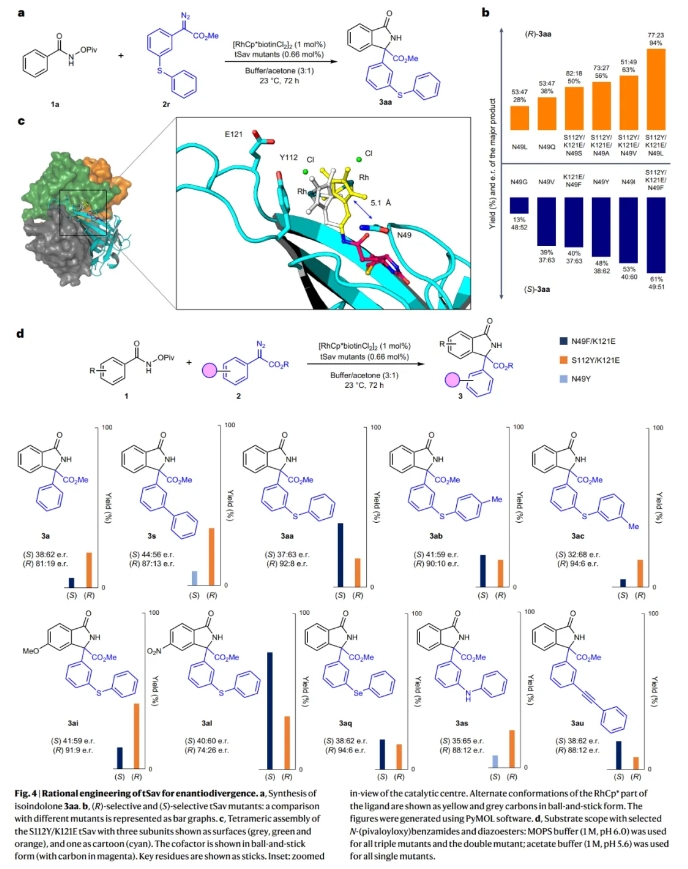
(图片来源:Nat. Synth.)
为了探索反应中的C-H活化过程的可逆性,作者以芳环被氘代的N-(特戊酰氧基)苯甲酰胺底物,在不存在重氮突变体的条件下,使用S112Y/K121E和N49Y在最优反应条件下反应48小时后并未在邻位观察到氢原子的引入。由此表明C-H活化步骤是不可逆的,这与之前的有机金属催化过程相一致(Fig. 5a)。
基于上述实验结果,作者提出了此转化可能的反应机理(Fig. 5b):首先,辅因子(中间体I)中的Rh(III)与酰胺的氮原子相互作用得到中间体II;随后通过协同金属化-去质子化进行C-H活化得到五元环铑中间体III并与重氮酯反应形成中间体IV;接下来,通过决定对映选择性的迁移插入得到六元环铑中间体V,并经历还原消除得到产物3a和Rh(I)。而Rh(I)可以被N-O键氧化为Rh(III),并通过质子去金属化过程促进活性催化中间体I的再生来完成催化循环。

(图片来源:Nat. Synth.)
总结
Prasenjit Bhaumik课题组和Debabrata Maiti课题组发展了链霉亲和素-生物素-铑(III)催化体系,以良好的对映选择性实现了手性异吲哚酮的合成。不同取代的N-(特戊酰氧基)苯甲酰胺和芳香重氮酯均具有良好的兼容性,实现了具有多种有用官能团(联苯、硫醚、硒醚、胺、烯烃和炔烃)的手性异吲哚酮的合成。机理研究表明该转化涉及一个导向的内球C-H活化过程以及随后的重氮插入过程。值得注意的是,作者通过设计得出的N49位点突变体可以有效实现相反的立体选择性,从而实现了对映发散合成。
文献详情:
Enantiodivergent synthesis of isoindolones catalysed by a Rh(III)-based artificial metalloenzyme
Prasun Mukherjee, Anjali Sairaman, Hirak Jyoti Deka, Shubhanshu Jain, Sandip Kumar Mishra, Sayan Roy, Prasenjit Bhaumik*,Debabrata Maiti*.
Nat. Synth., 2024
https://doi.org/10.1038/s44160-024-00533-5.
来源: 化学加
