
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
近日,美国科罗拉多州立大学(Colorado State University)Robert S. Paton和Andrew McNally课题组发展了一种将嘧啶转化为其它氮杂芳烃的新策略。通过将嘧啶转化为相应的N-芳基嘧啶盐,其经历裂解形成三碳单位的亚胺烯胺砌块,并应用于各种杂环形成反应中。这种连续的“有破有立”即解构-重建策略使嘧啶骨架更多样化,并能够获得其它的唑类杂环。利用此方法可以在复杂分子上形成杂环,从而产生用其它方法难以获得的类似物。相关成果发表在Nature上,文章链接DOI:10.1038/s41586-024-07474-1。
(图片来源:Nature)
正文
在药物的构效关系(SAR)研究中,修饰候选化合物的结构可以优化其在药物和农用化学品开发中的理化性质。然而,到目前为止,只有少数几种合成策略适用于小分子候选物中经常出现的氮杂芳烃的修饰。最近,美国科罗拉多州立大学Robert S. Paton和Andrew McNally课题组发展了将嘧啶转化为相应的N-芳基嘧啶盐,并经历连续的解构-重建策略实现了一系列取代嘧啶和唑类杂环的合成(Fig. 1)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
图片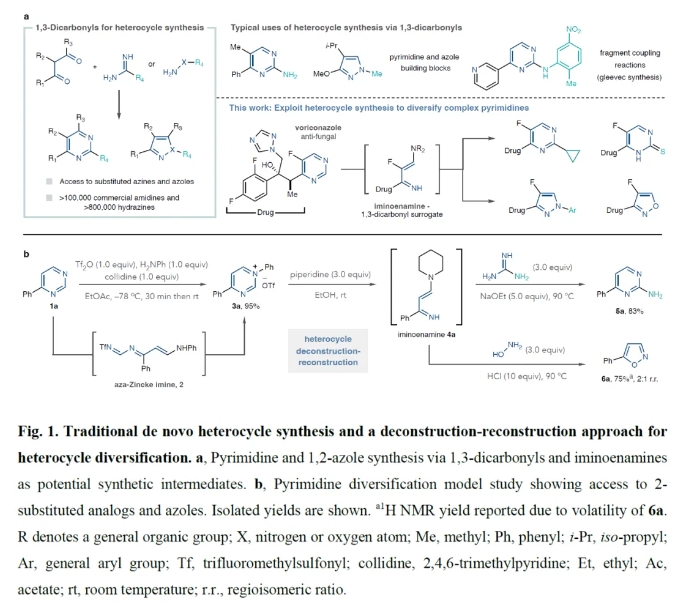
(图片来源:Nature)
Fig. 2展示了通过开环-关环反应过程合成嘧啶盐,并通过解构-重建方法将嘧啶转化为2-取代嘧啶和1,2-唑类的例子。在合成嘧啶盐的过程中,一系列不同取代的嘧啶,包括4-单取代嘧啶、5-单取代嘧啶、4,5-二取代嘧啶均可兼容此转化(3b-3g, 58-92%)。遗憾的是,喹唑啉类化合物在成盐过程中并不成功。反应中其它的不能实现转化的底物包括4-卤嘧啶,4-甲基嘧啶和2-取代的嘧啶。在这些情况中或者产率较低或者存在竞争性副反应。在解构-重建过程中,所形成的的亚胺烯胺中间体可以分别与脲(5b, 25%)、硫脲(5c, 93%)、三氟甲基乙脒(5f, 75%)等反应得到相应的2-取代的嘧啶。此外,一系列复杂生物活性分子片段同样可以兼容,以38-75%的产率得到相应的2-取代的嘧啶。值得注意的是,利用此策略还可以将嘧啶转化为唑类化合物6b-6g(40-90%)。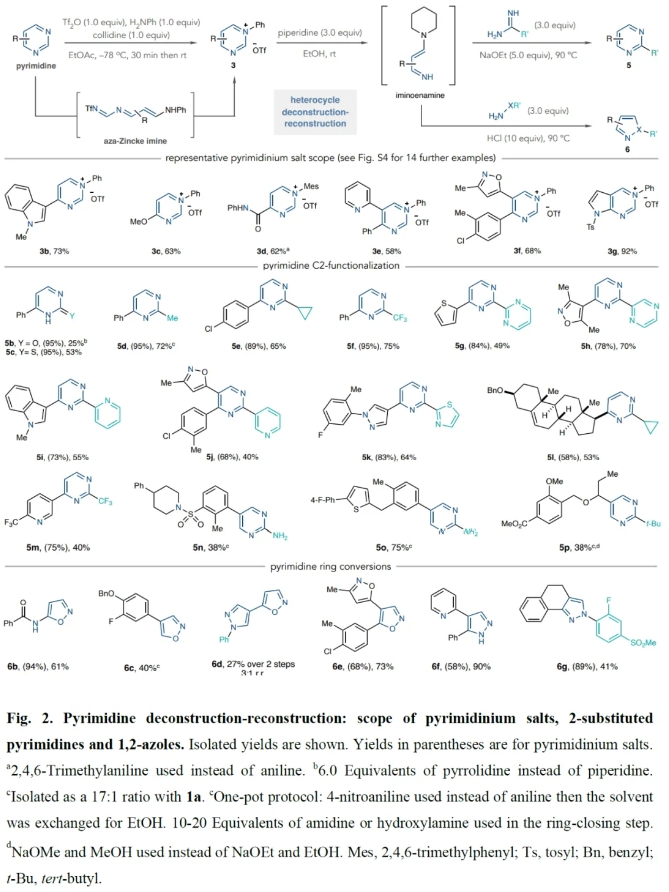
(图片来源:Nature)
为了进一步证明此转化的实用性,作者尝试利用此解构-重建策略实现生物活性分子的修饰(Fig. 3)。首先,作者利用一锅法策略将杀菌剂fenarimol (1b)转化为相应的氨基、环丙基和苯基衍生物5q-5s(28-38%)(Fig. 3a)。随后,为了探索更具挑战性的含嘧啶结构的兼容性,作者选择一种治疗黑色素瘤的小分子药物dabrafenib作为底物进行研究(Fig. 3b)。当利用一锅法策略实现嘧啶盐的原位形成后,作者分别使用胍、环丙基乙脒和三氟甲基乙脒与其反应可以分别得到2号位氨基化、环丙基化和三氟甲基化的产物5t (64%), 5u (61%), 5v (23%)。此外,将亚胺烯胺中间体4b进行断裂并分别经历与肼和羟胺的环化可以以47-86%的产率得到相应的唑类产物6h-6j。值得注意的是,利用吡啶盐3h的断裂以及NCS氯化和与环丙基乙脒的环化,可以以52%的产率得到双官能团化嘧啶产物5w。
(图片来源:Nature)
接下来,作者尝试利用所发展的解构-重建方法将嘧啶转化为吡啶(Fig. 4)。在嘧啶盐形成后,使用商业可得的乙酰基三甲基硅在NH4OAc和AcOH存在下95 oC反应可以以38-72%的产率得到相应的吡啶产物7a-7d。利用此转化可以实现形式上的C原子取代嘧啶N原子,这在SAR研究中起着至关重要的作用。此外,Fig. 4b展示了利用不同的亲核试剂实现了在C5或C2位引入取代基,以61-72%的产率得到相应的C5或C2位取代的吡啶产物7e-7h。
(图片来源:Nature)
最后,作者通过计算对嘧啶的开环机理进行了研究(Fig. 5)。Fig. 5a展示了1a与Tf2O和苯胺反应的计算势能面。1a和Tf2O之间的反应是放热的(-4.3 kcal mol-1),并通过TS-Act (ΔG‡ 12.6 kcal mol-1)一步进行。苯胺随后与NTf-嘧啶离子加成,且在C6 (ΔG‡ 15.1 kcal mol-1,通过TS-Add-C6)的亲核进攻要优于C2 (ΔG‡ 15.9 kcal mol-1,通过TS-Add-C2),因此在-78 oC时的动力学选择性为8:1。这些过渡结构(TS)的能量反映了电子Fukui (f +)指数,表明C6比C2 (0.15 vs 0.10)更具亲电性。虽然C6-加合物需要吸收8.9 kcal/mol热量来形成,但铵类物种的脱质子反应是一个无需活化能的反应过程,从而产生稳定的中间体Int-I (ΔG -20.7 kcal mol-1),这表明碱驱动了这一过程的发生。接下来,Int-I经历开环(ΔG‡ 10.7 kcal mol-1通过TS-II)形成aza-Zincke亚胺中间体Int-II。紧接着,Int-II通过互变异构异构化为Int-III,且在TS-IV (ΔG‡ 18.1 kcal mol-1,相对于Int-II)闭环之前以二氢嘧啶的形式形成Int-IV衍生物(ΔG -4.6 kcal mol-1)。由于关环过程需要最大的反应能垒,因此TS-IV的形成为决速步骤。最后,与生成的-NHTf阴离子产生一个稳定的离子对(Int-V),并通过随后的阴离子复分解得到产物3a(总体ΔG‡ -34.5 kcal mol-1)。此外,作者通过计算得出嘧啶取代基的大小和电子性质会显著影响产物形成的能垒(Fig. 5b)。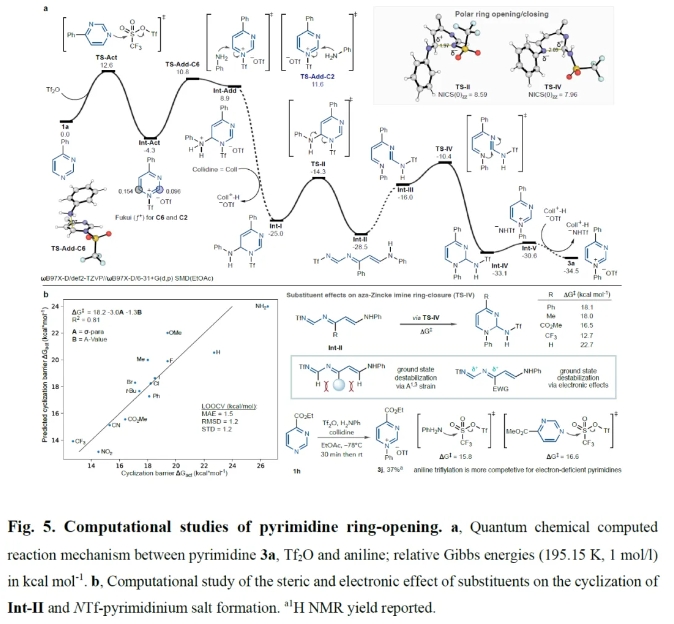
(图片来源:Nature)
总结
Robert S. Paton和Andrew McNally课题组通过将嘧啶转化为相应的N-芳基嘧啶盐,裂解形成三碳单位的亚胺烯胺砌块,并通过解构-重建策略实现了嘧啶骨架的多样性转化,发展了一种将嘧啶转化为各其它氮杂芳烃的新策略。此外,此策略还可以将嘧啶转化为其它方法难以获得的唑类杂环。此解构-重建反应的发展为其它杂环的构建提供了新的思路。
文献详情:
A deconstruction-reconstruction strategy for pyrimidine diversification.
Benjamin J. H. Uhlenbruck, Celena M. Josephitis, Louis de Lescure, Robert S. Paton*, Andrew McNally*.
Nature, 2024
https://doi.org/10.1038/s41586-024-07474-1.
来源: 化学加
