
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 HyperAI超神经
HyperAI超神经 
自 2020 年被 Nature Methods 评选为年度技术以来,「空间转录组学」已成为当今生命科学领域最炙手可热的革命性技术之一。简单来说,该技术能够获取组织空间信息和转录组数据,从时间和空间维度精准解析组织内基因表达模式,以及细胞类群的空间位置关系等生物学特征,在疾病研究、生长发育、器官结构和物种演化等领域的研究中均具有极高价值。
随着空间转录组学在学术科研领域持续火爆,如 10x Visium、Slide-seq 以及 Stereo-seq 等空间转录组学技术也如泉涌而出,这些最新成果和进展正彻底地改变人类对组织内基因表达模式的研究。然而,仅仅获得组织内特定物理坐标的基因表达谱,还无法全面了解生物系统的复杂性,穷究其理,必须辨别组织内每个空间点的生物学身份。
目前,基于机器学习的方法已被广泛应用于识别空间点的群集,并使用标记基因解释其生物学身份。但这些方法通常因为缺乏与群集内已知结构建立明确联系的能力而受限。此外,手动注释常被用作辅助识别已知结构,但该方法常受限于研究人员的专业知识和主观判断,且无法应用于大规模分析当中。
针对上述挑战,复旦大学脑科学研究院诸颖团队近日在「Nature Communications」上发表了题为「Pianno: a probabilistic framework automating semantic annotation for spatial transcriptomics」的研究成果。研究团队借鉴了计算机视觉中的「语义分割」思想,提出了「空间转录组语义注释」概念,并开发了空间转录组语义注释工具 Pianno,能够为组织内的空间点自动定义结构或细胞类型,从而结合来自多个维度的信息,加强对复杂生物系统的解释。
研究亮点:
Pianno 具有独特的自动标记模式,适用于各种空间转录组学技术生成的数据
与最先进的空间聚类方法相比,Pianno 展现出了卓越的性能,为空间转录组学数据提供了新的视角

数据集:公开数据,严谨计算
该研究所用的数据集主要为公共数据集,来自不同的空间技术平台,具体包括了人类背外侧前额叶皮质数据集 dlPFC、成年小鼠大脑半球冠状切片数据集 Stereo-seq、小鼠海马体预处理数据集 Slide-seqV2、人类胰腺导管腺癌数据集 ST、人类乳腺癌数据集 Visium、小鼠初级视觉皮层数据集 scRNA-seq、多个人类皮质区域的 snRNA-seq 数据集、小鼠嗅球 (olfactory bulb) 的 DAPI 染色图像等。
研究中,为避免降噪、平滑、锐化等图像处理技术对生物原始特征造成破坏,研究团队基于原始计数构建了贝叶斯分类器 (Bayesian classifier) 以微调初始注释。同时,研究团队应用了高阶马尔科夫随机场 (Markov random field, MRF) 先验模型。在空间转录组学背景下,由于必须共同考虑每个位点的基因表达和空间位置,研究团队还采用了空间泊松点过程 (spatial Poisson point process, sPPP) 模型。
Pianno:创新的自动化空间转录组语义注释新工具
研究团队提出了一个基于贝叶斯框架的新工具 Pianno,该工具结合了马尔可夫随机场 (MRF) 与空间泊松点过程 (sPPP),充分利用了 sPPP 建模 RNA-seq 计数数据分布的能力,同时考虑了空间点的位置信息,可以使用预定义的标记基因列表,自动化地注释空间转录组数据中每个点的生物身份。

Pianno 框架
Pianno 输入的空间转录组数据由空间坐标 (Spatial coordinates)、初始标记基因列表 (Initial marker list) 和原始基因计数 (Spatial raw counts) 组成,每个模式至少提供一个已知的标记。
注释过程由初始分割步骤 (initial segmentation step) 和精细化步骤 (refinement step) 构成:
在初始分割步骤中,每个基因的空间表达被转换为灰度图像 (grayscale image)。对于每个目标模式 (pattern),通过聚合与该模式相关的标记基因的灰度图像来创建模式图像 (pattern image),然后确定每种模式的额外候选标记基因来更新初始标记列表 (Updated marker list)。考虑到它们在最初注释的结构中的独特表达模式,更新后的标记列表将会被整合到后续的精细化步骤中。
在精细化步骤中,构建一个贝叶斯分类器 (Bayesian classifier) 来评估每个空间点属于不同模式的后验概率 (posterior probability),然后根据后验概率更新注释。
Pianno 提供两种更新注释的方法:
对于语义标注中的连续模式,建议将概率分布作为模式图像,返回给模式检测器 (Pattern detector) 进行更新标注;
对于分散或尖锐的图像模式,建议根据概率值直接更新标签,因为它可以保留详细信息。
总的来说,Pianno 简化了注释过程,同时采用启发式方法 (heuristic approach) 使用初始单个标记基因来识别额外的标记基因,可以最大限度减少对已知标记数量的输入。
研究结果:性能卓越、适用性强
在本次研究中,研究团队对 Pianno 的性能、准确性、适应性等进行了验证,并通过与现有方法进行比较,进一步论证了 Pianno 的能力。
在与基于聚类的工具在解剖结构注释的比较中,研究团队使用 dlPFC 数据集中的 12 个样本对 Pianno 的性能进行了评估,并与另一种基于标记但无空间信息的注释方法 CellAssign 进行了比较。另外,评估过程中还考虑了无监督聚类方法 Leiden 算法,以及 5 种空间聚类方法 (SpaGCN、SEDR、BayesSpace、DeepST 和 STAGATE)。

Pianno 在皮层结构重建中的性能表现评估
评估发现,Pianno 的性能与经验丰富的研究员在基于形态特征和标记进行的手工注释,达成了最高一致性,在 12 个样本中,有 11 个样本优于其他测试方法。
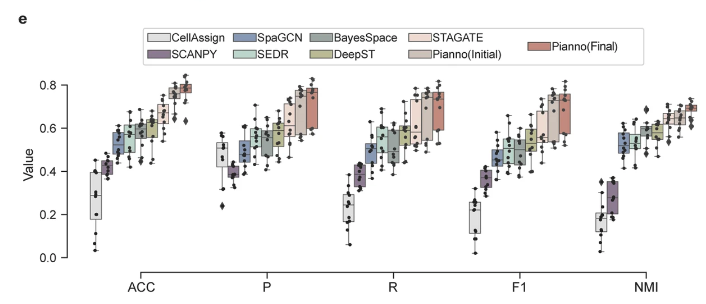
多个指标评估结果
此外,研究团队还通过其他分类指标,如准确率 (ACC)、宏平均精确度 (macro-averaging precision, P)、宏平均召回 (macro-averaging recall, R)、宏平均 F1 分数 (F1) 和归一化互信息 (normalized mutual information, NMI),进一步全面评估了 Pianno 的优越性能,如上图 e 所示,Pianno 相关指标均位于较高水准。
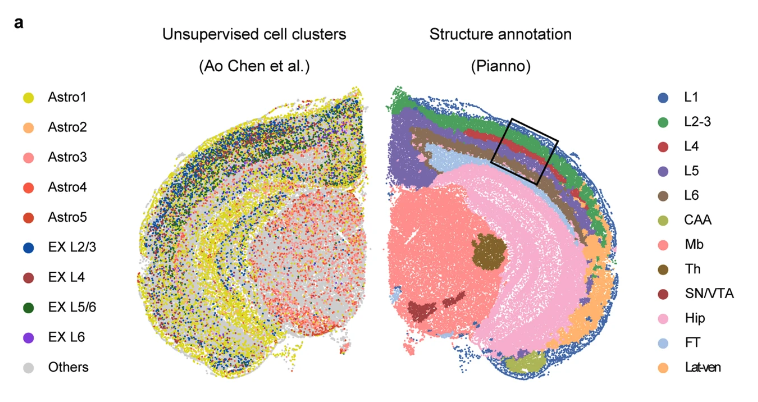
Pianno在小鼠皮质内细胞类型注释性能基准测试
随后,研究团队评估了 Pianno 预测细胞类型空间分布的能力。在本轮验证中,研究团队使用了成年小鼠半脑冠状切片的 Stereo-seq 数据集,并将结果与通过不同策略推断的细胞类型分布进行了比较,包括细胞分割后进行无监督聚类,以及 3 种基于空间和单细胞转录组学整合的空间去卷积 (deconvolution) 工具。
研究发现,Pianno 对兴奋性神经元亚型分布的预测,显示出与 Tangram 和 RCTD 相当的模式,与它们在各层已知位置上表现出高度的一致性。总的来讲,该结果证明了 Pianno 在预测空间数据集中复杂的细胞类型分布方面,具有很好的稳健性和准确性,尤其是在无监督方法遇到挑战的情况下。
然后,研究团队进一步评估了 Pianno 在不同平台的空间转录组数据中注释各种形状结构的性能,并与 STAGATE 进行了比较。
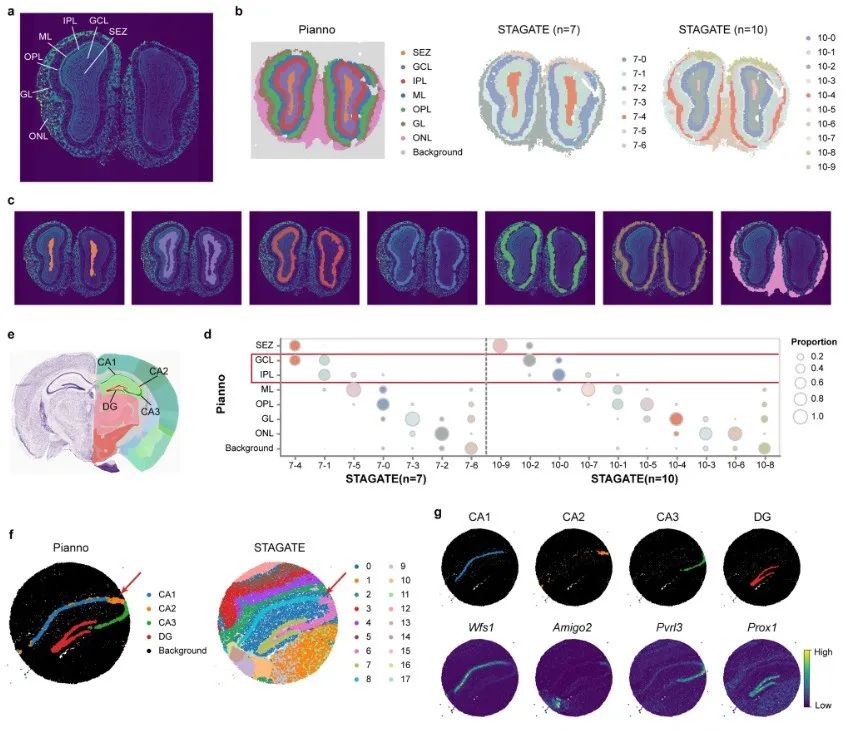
Pianno 在不同平台上注释各种形状结构的性能表现
研究团队使用 Pianno 对小鼠嗅球的 Stereo-seq 数据集中的解剖结构进行了注释,该数据集包含了 10,747 个空间点,涵盖组织覆盖区域和背景区域。
Pianno 能够在几分钟内同时完成背景去除和结构标注。相比之下,当将聚类数设置为结构数时,STAGATE 则无法识别出所有解剖结构对应的聚类。
研究团队还针对肿瘤微环境呈现出的高度异质性,评估了 Pianno 在注释复杂且分散结构组织方面的表现。本轮测试分析了 2 个人类胰腺导管腺癌样本和 2 个乳腺癌样本的微环境。
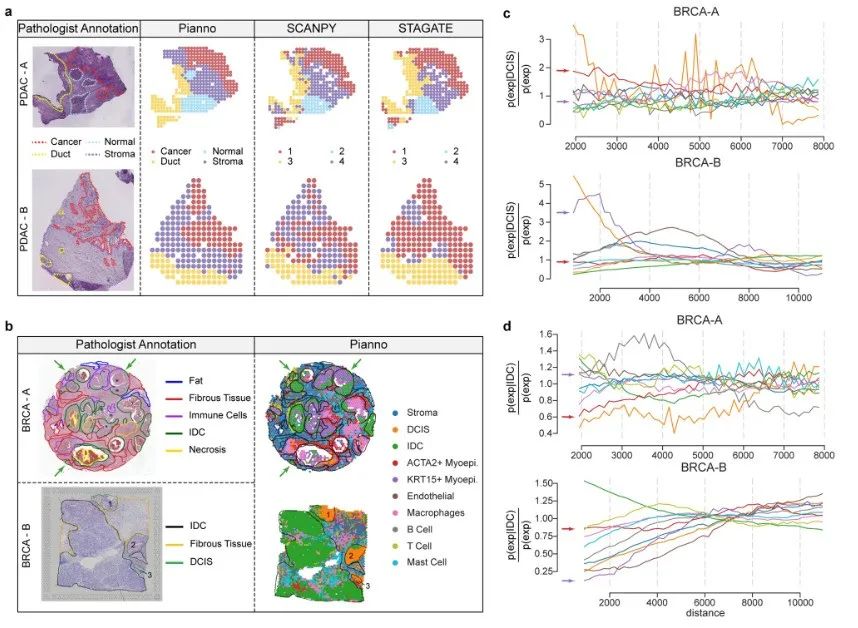
肿瘤微环境注释
总体而言,Pianno 表现出与专业病理学家手工标注一致的水平,证明了其在注释不规则的复杂结构时,特别是在异质性肿瘤微环境中,具有极大潜力。这对病理学家在理解肿瘤生物学的复杂性方面提供了宝贵的帮助,并有望为提供个性化治疗策略找到新思路。
人工智能与复杂生物学结合大有可为
根据复旦大学脑科学研究院报道,目前该研究项目已得到国家重点研发计划「生物与信息融合 (BT 与 IT 融合)」重点专项、科技创新 2030 —「脑科学与类脑研究」重大项目、国家自然科学基金、上海市科技重大专项和张江实验室等基金的资助。
据了解,复旦大学脑科学研究院成立于 2006 年 4 月,是复旦大学全校性的神经科学研究实体机构、教育部「985 工程」二期重点建设的科技创新平台之一,与医学神经生物学国家重点实验室为「两位一体」建设项目。
从建院至今,复旦大学脑科学研究院已经结下累累硕果。该院多次面向国际和国家重大需求,承担重大科研项目,产出重要研究成果。据其官网披露,该院研究人员已主持参与了一系列重大科研项目,包括科技部「973 计划」、「863 计划」、科技创新 2030 「脑科学与类脑研究」、国家重点研发计划、国家科技重大专项「重大新药创制」等等。
其实,除了复旦大学脑科学研究院外,目前已有不少实验室、企业也开始关注到空间转录组技术。
例如,中国科学院数学与系统科学研究院张世华团队开发了 STA-系列工具。2022 年,该团队发布了适应于不同空间转录组技术、不同生物组织的生物组织空间亚结构识别的人工智能工具 STAGATE。进入 2023 年后,该团队又围绕空间转录组技术发布了多项成果
针对来自不同技术、不同发育时间点、不同疾病条件的生物组织多切片空间转录组数据建立了整合分析新工具 STAligner。
基于深度学习显著图的空间域特异可变基因识别方法 STAMarker,同时实现了空间域识别和对应的空间可变基因识别,有望为细粒度分析空间转录组数据提供有效方法。
与中国科学院北京基因组研究所(国家生物信息中心)杨运桂、蔡军团队合作,绘制了地中海涡虫再生过程中的三维空间转录组图谱 STAPR,系统鉴定了多个再生关键调控因子。

ENGEP 增强空间转录组学数据
华中师范大学数学与统计学学院张晓飞教授课题组开发了一款名为 ENGEP 的计算方法,利用 k近邻加权回归和集成学习策略,能够准确预测空间转录组中未测基因的表达。此外,ENGEP 还能够准确预测空间未测基因的表达模式,对增强空间转录组学数据具有重要意义。
毫无疑问,AI 在空间转录学、乃至生物学领域的赋能不仅提高了研究效率,同时还为科研难点提供了新的解题思路。正如该论文在讨论部分指出的 Pianno 所带来的价值——可能取代现有劳动密集型的人工注释,以自动化的方式提供高效、精准、低成本的形式为空间转录组学带来变革,也将推动生物学新发展。
参考资料:
1. https://news.fudan.edu.cn/2024/0407/c2474a139894/page.htm
2. https://bfse.cas.cn/sxyqyjc/kyjz/202311/t20231110_4985132.html
3. https://kjc.ccnu.edu.cn/info/1009/3744.htm
来源: HyperAI超神经
