
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 人染色体
人染色体 人染色体
第五章
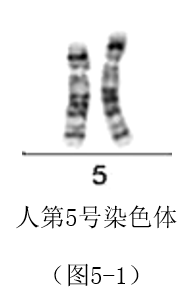
5 号染色体大小、形态、带纹与4号染色体相似;其长180.915Mb,编码894个基因(信息参考:Ensembl GRCh37,release 109 - Feb 2023 © EMBL-EBI),基因密度比值约为4.94。
第5 号染色体短臂(p15)缺失,引起的疾病称猫叫综合征(Cri-du-Chat综合征,OMIM#123450),又名5p–综合征,为最常见的染色体缺失综合征,因婴儿期啼哭声似猫叫而得名,其原因在于5p15部分有末端或中间片段缺失,缺失片段包括了喉肌发育的基因,导致患儿的喉部发育不良或未分化。这种综合征约占所有智力发育迟缓患者的1%。患儿头小,圆脸,眼距增宽,眼角下斜;生长、智力发育落后,伴先天性心脏病(50%), 多能存活到成年。发病率约为1/50000,女患者多于男患者。5P染色体缺失片段的断裂点和范围在不同的患者中有所不同,但在所有有表型的患者中缺失的关键区域被鉴定为5P15。许多临床发现该综合征似乎是由于5p15片段内的一个或多个基因单倍剂量造成的,智力发育迟滞的程度通常与缺失的片段大小有关。
家族性腺瘤性息肉病(familial adenomatous polyposis, FAP, OMIM #175100))是一种常染色体显性的癌前病变,通常会进展为恶性腺瘤。FAP患病率约为2~3人/10万人,其致病基因APC(一种抑制肿瘤发生的基因)位于5q21,该基因编码一种肿瘤抑制蛋白,该蛋白直接或间接调节转录、细胞粘附、微管细胞骨架、细胞迁移、凋亡和细胞增殖。APC的两个等位基因都必须失活才能形成肿瘤,肿瘤是一种体细胞遗传病,这种体细胞功能的丧失是由于杂合子缺失、基因突变、转录失活以及遗传突变等位基因的显性负作用(基因的功能是显性,但由于功能丢失不能起作用)。尽管APC的功能缺失会导致受影响的细胞增生异常,但这些细胞还必须获得体细胞内其它的基因改变才能发展为癌症。这种发展的特点是细胞遗传学不稳定,导致大量染色体片段丢失,从而导致杂合子丢失。与这一发展相关的特异性基因改变包括Ki-Ras或N-Ras癌基因的激活,18q上肿瘤抑制基因DCC的失活,17P上的TP53基因的失活,以及甲基化的改变导致肿瘤抑制基因的转录沉默。随着细胞积累突变。它们变得越来越肿瘤化,最终形成侵袭性和转移性癌(见图5-2)。

家族性腺瘤性息肉病(FAP),临床表现为整个结直肠(大肠)布满大小不一的腺瘤(数百到数千个)。家族成员多在15岁前后出现息肉,初起时息肉为数不多,随着年龄增长而增多。家族性腺瘤性息肉病如不及时治疗,终将发生癌变。有家族性腺瘤性息肉病家族史的个体,18岁以后,每年检查(特别是男性青年),及时手术切除癌变组织,以防癌细胞转移。80%以上的散发性结肠肿瘤伴有体细胞APC突变。
家族性腺瘤性息肉病的诊断并不困难。有家族史,青年期即可发病,有腹部隐痛、腹泻、黏液血便等结肠息肉的症状,结肠镜下结肠内有数百枚息肉,病理检查为腺瘤性息肉为主,即可诊断本病。
大约2%到4%的结肠癌病例是由Lynch综合症发展而来,Lynch综合症属常染色体显性遗传病(AD),该病伴少量腺瘤性息肉,在青春期以前,腺瘤性息肉为良性瘤。与家族性腺瘤性息肉病(Familial Adenomatous Polyposis,FAP,所见的数百至数千腺瘤性息肉相反,息肉的数目一般很少,然而,Lynch综合症息肉有很高的恶性转化潜能。Lynch综合症最常见的杂合子基因突变(基因处于杂合状态,其中的一个正常等位基因发生了丢失或突变)在一生中大约有80%的风险发展成结肠癌;女性风险略低,为70%,但有约40%的风险患子宫内膜癌;胆管癌、泌尿道癌和卵巢癌的风险也增加10%到20%,皮肤皮脂腺肿瘤可能是Lynch综合症的第一个临床表现。
结肠癌是常见的消化道恶性肿瘤,涉及多个基因的变异(基因缺失、突变等),以40~50岁人群发病率最高,男女之比为2~3:1。慢性结肠炎患者、结肠息肉患者、男性肥胖者等为易感人群。
结肠癌是多基因和环境共同作用引起的疾病,除遗传基因变异外,环境引起的原因主要与高脂肪和低纤维素饮食有关,注意饮食低脂肪高纤维的摄取,有助于降低结肠癌的发生。
脊髓性肌萎缩症(spinal muscular atrophy,SMA)又称脊肌萎缩症。系指一类由于以脊髓前角细胞为主的变性导致肌无力和肌萎缩的疾病。首先由Werdnig(1891)和Hoffmann(1893)报道,故又称Werdnig-Hoffmann病。根据起病年龄和病变程度可将本病分为4型:Ⅰ~Ⅲ型称为儿童型脊髓性肌萎缩症,属于常染色体隐性遗传病(AR),致病基因SMN位于5q13,群体发病率为1/6000~1/10000,是婴儿期最常见的致死性遗传病。儿童型脊髓性肌萎缩症SMN基因异常,多为外显子7、8缺失,男女均可患病,一般男性多于女性。
20~30岁以上起病的SMA,归为第Ⅳ型,可呈常染色体隐性(AR)、显性(AD)和X连锁隐性(XR)等不同遗传方式,其群体发病率约为0.32/10000。
各型区别根据起病年龄、病情进展速度、肌无力程度及存活时间长短而定。至今本病尚无特异的有效治疗,主要治疗措施为预防或治疗严重肌无力产生的各种并发症,如肺炎、营养不良、骨骼畸形行动障碍和精神社会性问题等。
哮踹在人群中极为常见,研究发现,与“哮喘”有关的基因,在5号染色体上不是一两个,而是好多个!它们各自分别影响哮喘的发生,但各自的作用微小(在其它染色体上,也分布着能导致哮喘的多个基因),但没有一个基因能够对哮喘的发生起到主要作用;相反,环境因素控制起这种疾病来,却显得举足轻重。调查发现,那些讲卫生勤洗手、洗澡的孩子,比那些很少洗手洗澡的孩子更易患这种病,经常接触哮喘致敏源的孩子对致敏物(抗原)产生了抗体,使得他们不易患病。多个基因参与生物性状控制的结果就是,在很多情况下,这些性状绝不会像单基因病-亨廷顿舞蹈症那样有一个基因异常则有病、无基因异常则无病,而是有着所谓的连续性的数量性状即不同患者的病情严重程度不一致。多基因遗传病涉及人群60%以上的个体,其种类虽然没有单基因遗传病多,但涉及面之广泛,是单基因病不能比拟的,如前面提及的阿尔兹海默病、结肠癌、肺癌均为多基因病。
待续
来源: 医学遗传学教材,网络
