
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助

分子轨道理论又称分子轨道法(Molecular Orbital Theory)或MO法,1932年由美国化学家密立根(R.S.Mulliken)及德国物理学家洪特(F.Hund)提出。是现代共价键理论之一。它的要点是:从分子的整体性来讨论分子的结构,认为原子形成分子后,电子不再属于个别的原子轨道,而是属于整个分子的分子轨道,分子轨道是多中心的;分子轨道由原子轨道组合而成,形成分子轨道时遵从能量近似原则、对称性一致(匹配)原则、最大重叠原则,即通常说的“成键三原则”;在分子中电子填充分子轨道的原则也服从能量最低原理、泡利不相容原理和洪特规则。1
分子轨道的历史1927年,我们所熟知的价键理论被提出,随后,受到洪特,马利肯,斯莱德和John Lennard-Jones的影响,分子轨道开始产生。因此,在最初的时候,分子轨道理论被称为洪特-马利肯理论。而“轨道”一词的概念则是在1932年首先被马利肯提出。
到了1933年,分子轨道理论已经被广泛的接受,并且被认为是一个有效而且有用的理论。事实上,根据德国物理化学家休克尔的描述,第一篇使用分子轨道理论的文献是由莱纳德琼斯发表于1929年。而第一个使用分子轨道理论的定量计算文献则是在1938年由库尔森发表的使用自洽场理论解决氢分子的电子波函数的工作。
到1950年,分子轨道彻底被定义为自洽场哈密顿算符的本征函数,这就是分子轨道理论发展成为一个严谨科学理论的标志。HF方法(Hartree-Fock method)是分子轨道理论的一种比较严谨的处理方法,尽管在一开始,HF方法是用来计算原子的电子结构的一种方法,但是在分子计算当中,分子轨道按照原子轨道的一组基集被拓展,发展出罗特汉方程,以此为基础,又发展出了各种各样的从头算量子化学计算方法。与此同时,分子轨道理论也被应用在了一种采用了更多近似方法的半经验计算当中,被称为半经验量子化学计算方法。2
理论简介一种化学键理论,是原子轨道理论对分子的自然推广。其基本观点是:物理上存在单个电子的自身行为,只受分子中的原子核和其他电子平均场的作用,以及泡利不相容原理的制约;数学上则企图将难解的多电子运动方程简化为单电子方程处理。因此,分子轨道理论是一种以单电子近似为基础的化学键理论。描写单电子行为的波函数称轨道(或轨函),所对应的单电子能量称能级。对于任何分子,如果求得了它的系列分子轨道和能级,就可以像讨论原子结构那样讨论分子结构,并联系到分子性质的系统解释。有时,即便根据用粗糙的计算方案所得到的部分近似分子轨道和能级,也能分析出很有用处的定性结果。
轨道简介1、原子在形成分子时,所有电子都有贡献,分子中的电子不再从属于某个原子,而是在整个分子空间范围内运动。在分子中电子的空间运动状态可用相应的分子轨道波函数ψ(称为分子轨道)来描述。分子轨道和原子轨道的主要区别在于:
(1)在原子中,电子的运动只受1个原子核的作用,原子轨道是单核系统;而在分子中,电子则在所有原子核势场作用下运动,分子轨道是多核系统。
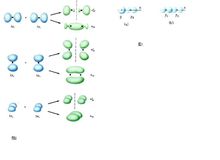 (2)原子轨道的名称用s、p、d…符号表示,而分子轨道的名称则相应地用σ、π、δ…符号表示。
(2)原子轨道的名称用s、p、d…符号表示,而分子轨道的名称则相应地用σ、π、δ…符号表示。
2、分子轨道可以由分子中原子轨道波函数的线性组合(linear combination of atomic orbitals,LCAO)而得到。有几个原子轨道就可以可组合成几个分子轨道,其中有一部分分子轨道分别由对称性匹配的两个原子轨道叠加而成,两核间电子的概率密度增大,其能量较原来的原子轨道能量低,有利于成键,称为成键分子轨道(bonding molecular orbital),如σ、π轨道(轴对称轨道);同时这些对称性匹配的两个原子轨道也会相减形成另一种分子轨道,结果是两核间电子的概率密度很小,其能量较原来的原子轨道能量高,不利于成键,称为反键分子轨道(antibonding molecular orbital),如 σ*、π* 轨道(镜面对称轨道,反键轨道的符号上常加“*”以与成键轨道区别)。还有一种特殊的情况是由于组成分子轨道的原子轨道的空间对称性不匹配,原子轨道没有有效重叠,组合得到的分子轨道的能量跟组合前的原子轨道能量没有明显差别,所得的分子轨道叫做非键分子轨道。
3、电子在分子轨道中的排布也遵守原子轨道电子排布的同样原则,即Pauli不相容原理、能量最低原理和Hund规则。具体排布时,应先知道分子轨道的能级顺序。当前这个顺序主要借助于分子光谱实验来确定。
线性组合原则原子轨道组合形成分子轨道时所遵从的能量近似原则、对称性匹配原则和轨道最大重叠原则称为成键三原则。1
对称性匹配原则只有对称性匹配的原子轨道才能组合成分子轨道,这称为对称性匹配原则。原子轨道有s、p、d等各种类型,从它们的角度分布函数的几何图形可以看出,它们对于某些点、线、面等有着不同的空间对称性。对称性是否匹配,可根据两个原子轨道的角度分布图中波瓣的正、负号对于键轴(设为x轴)或对于含键轴的某一平面的对称性决定。
能量近似原则在对称性匹配的原子轨道中,只有能量相近的原子轨道才能组合成有效的分子轨道,而且能量愈相近愈好,这称为能量近似原则。
轨道最大重叠原则对称性匹配的两个原子轨道进行线性组合时,其重叠程度愈大,则组合成的分子轨道的能量愈低,所形成的化学键愈牢固,这称为轨道最大重叠原则。在上述三条原则中,对称性匹配原则是首要的,它决定原子轨道有无组合成分子轨道的可能性。能量近似原则和轨道最大重叠原则是在符合对称性匹配原则的前提下,决定分子轨道组合效率的问题。
轨道能量决定于组成原子轨道的类型和原子轨道间的重叠,例如σg1s和σu1s比σg2s低得多,这是由于原子轨道1s的能量比2s的低得多。同理,因为除氢原子外,2s能量显著低于2p的能量,故σg2s比σg2p能量低。另外,只要核间距不很小,两个2s轨道或两个2pz轨道之间的重叠比两个2py或2px之间的重叠大得多,因此成键和反键π轨道间的能量差比对应的σ轨道的差小。根据这种论述,表2中所列分子轨道次序可预料为:σg1s
