
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助

Paternò-Bǜchi(P-B)反应,即羰基-烯的[2+2]光环化加成反应,利用其特殊的区域及立体选择性可以合成一些结构精巧的取代氧杂环丁烷。随着P-B反应在有机合成中越来越广泛的应用,人们对P-B反应的区域选择性的研究也越来越深入。在P-B反应发现之初,人们一直用“最稳定的双自由基规则”解释其区域选择性,这一规则对有些体系却不适用,近年来有人开始运用“自旋化学”理论来解释区域选择性。
简介P-B反应是羰基化合物与烯烃的[2+ 2]光环化加成反应,该反应自从50年代初建立以来,已成为构建氧杂环丁烷的一种重要的合成方法。它可用来合成一些热或其它化学方法难以得到的结构精巧的取代氧杂环丁烷。然后利用氧杂环丁烷在不同反应条件下的不同反应,可以合成一些结构精巧的化合物。
现在被普遍接受的P-B反应历程为:首先羰基化合物吸收光子被激发到单重态,单重态可以直接与烯组分反应,也可以经历系间窜跃形成激发三重态,然后再与烯组分反应,生成三重态的1,4-双自由基,再经历系间窜跃到单重态的1,4-双自由基,关环生成氧杂环丁烷或发生β断裂回到基态的反应物。
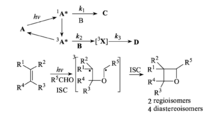
其中三重态可以通过猝灭去活化,这样就可以研究三重态的化学行为。在单重态反应途径中,进攻烯组分产生的单重态双自由基中间体的寿命非常短,区域选择性被自由基-自由基重合的优势几何学控制;而三重态反应途径中产生三重态的1,n-双自由基,区域选择性与这种双自由基中间体的稳定性有关。因此研究三重态的1,4-双自由基性质是研究P-B反应区域选择性的重要手段。1
特点P-B反应有着广泛的羰基-烯反应体系。对于烯组分,除简单烯烃外,还有α位被杂原子取代的衍生物,如烯醚、环状的乙烯基碳酸酯、氧杂环烯及其衍生物(如2,3-二氢呋喃或吡喃体系)、硅烯醚、硫烯醚、烯胺以及其它的一些杂环化合物及其衍生物;炔类化合物也可以作为有效的烯组分与羰基化合物发生P-B反应生成氧杂环丁烯,可以经开环形成烯酮,也可以进一步与另一分子羰基化合物反应。对于羰基组分,可以为脂肪族或芳香族的醛或酮,也可以是芳香羧酸酯。另外苯腈与烯烃也可以发生P-B反应。当一个分子中同时含有羰基和烯组分时,可以发生分子内的P-B反应,熵变应该使分子内的P-B反应比分子间的反应更容易。但是Norrish typeⅡ光化学反应和抽氢反应的竞争使分子内P-B反应发生的可能性很小。在发生P-B反应的同时,还可能伴随着电子转移、能量转移和抽氢反应等副反应。随着有机合成的发展,P-B反应的立体及区域选择性越来越受到有机化学家们的关注。现在对于P-B反应的立体选择性已经有了比较系统的报道。P-B反应的立体选择性包括由不对称羰基组分引起的简单的立体选择性和由羰基或烯烃的手性中心诱导产生的“诱导非对映选择性。1
P-B反应的区域选择性的研究机理1、最稳定的双自由基规则
过去,人们大多按照“最稳定的双自由基规则”来讨论P-B反应的区域选择性,这一规则在多数体系是简单有效的。这些P-B反应体系往往仅能得到一种或一种占绝对优势的区域产物,这是由于在反应过程中形成一种非常稳定的双自由基中间体,这种最稳定的双自由基中间体形成的产物就是优势产物。
D'Auria小组通过研究发现,2,3-二氢呋喃、呋喃、2-呋喃甲醇发生P-B反应时,其区域选择性(进攻呋喃环上的α-C还是β-C)可以由HOMO的原子系数解释;单取代呋喃的区域选择性(对呋喃环上双键的选择)可以由形成的双自由基中间体的稳定性解释,稳定的中间体形成优势产物。
例如2,3-二氢呋喃和呋喃分别与苯甲醛发生P-B反应时的区域选择性正好相反。当苯甲醛与2,3-二氢呋喃反应时endo-3-烷氧基氧杂环丁烷为主要产物;但是,当苯甲醛与呋喃反应时形成exo--烷氧基氧杂环丁烷。
其原因是:羰基激发态的氧原子有亲电的自由基性质,因此氧原子的半充满的n轨道更倾向于进攻烯烃上电子云密度较高的位置。通过环烯烃的HOMO系数可以很容易推测出氧原子应该进攻二氢呋喃的3位和呋喃的2位分别形成三重态双自由基中间体DR-1和DR-2。另一方面,DR-2双自由基比氧原子进攻呋喃的3位形成的区域异构体稳定,这是因为在DR-2中的呋喃环上形成稳定的烯丙自由基体系。因此在呋喃及其衍生物的反应中形成唯一的exo-2-烷氧基氧杂环丁烷。
“最稳定的双自由基规则”忽略了电子激发态羰基与烯组分在反应过程中相互接近的几何学。事实上,以不同接近方式生成的双自由基在势能上存在差异,已有许多体系无法用这一规则解释。近几年,人们开始对一些体系的区域选择性提出新的解释。
2、Salem-Rowland规则
在有机光化学反应中常涉及自旋多重态,总反应的反应活性和选择性与自旋态有关,这被称为自旋化学。其主要特点就是自旋守恒原则,即只有当反应物与产物的自旋态相同时才能发生化学反应,当反应物转变成产物需要发生自旋态改变时是自旋禁阻的。但在实际反应中并不严格遵守这个规则,有时在反应过程中只有发生自旋态的改变才能形成产物,此时分子自旋态的改变完全决定反应的速率与选择性,这被称为双态反应(TSR)。
从三重态双自由基转化为闭壳层产物需要发生系间窜跃(ISC),这是一个自旋禁阻的过程,ISC的速率主要由旋轨耦合(SOC)控制。与其它相互作用如电子-核超精细耦合(HFC)和自旋-晶格弛豫(SLR)相比,SOC强烈地依赖于三重态双自由基的几何构型,被称为Salem-Rowland规则。其要点是:(1)增加两自旋原子之间的距离SOC降低。由于额外的键传递的相互作用,使得SOC不仅与两自由基中心的实际距离有关,而且与经过的键的数目(n- 1)有关;(2)角度守恒要求自由基中心的p轨道的轴线相互垂直;(3)SOC与相应的双自由基的离子性质成比例。1
区域选择性的近期研究进展当P-B反应生成两种稳定性相近的双自由基中间体时就可能得到两种区域产物。这样三重态的1,4-双自由基中间体的稳定性对区域选择性就不再起决定作用,从而使其它影响因素的作用显现出来。目前人们对区域选择性的报道大都集中于不对称取代呋喃衍生物和非环状的烯烃衍生物的P-B反应的区域选择性,但这些都是三重态双自由基的稳定性对区域选择性起决定作用,而对于影响区域选择性的其它影响因素(如底物浓度、激发态的性质、溶剂的极性和粘度、温度和取代基等等)的报道则很少,现在仅有的是不对称取代呋喃和嘧啶衍生物与芳香性羰基化合物反应的区域选择性的温度效应和取代基效应。1
本词条内容贡献者为:
黄伦先 - 副教授 - 西南大学
