
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 HyperAI超神经
HyperAI超神经 
分子逆折叠在药物和材料设计中起到关键作用,使得科学家能够合成具有理想结构的新分子。过去的研究大多集中于大分子或小分子的逆折叠,但却很少关注通用分子的逆折叠。
**构建统一的通用模型主要存在 3 大挑战:**① 单位差异:大分子一般将预定义的微结构作为基本单元,如氨基酸之于蛋白质,核苷酸之于 RNA;而小分子将原子作为基本单元;② 几何特征提取:不同研究在几何特征提取方面采用多种策略,如距离、角度和张量积,缺乏统一的特征化方法;③ 系统规模:小分子允许全局注意力机制来学习长期依赖关系,但这往往在大分子上行不通。
为了解决上述挑战,并进一步补充 RoseTTAFold All-Atom 和 AlphaFold 3 在分子结构预测上取得的进展,**来自西湖大学未来产业研究中心的团队提出了一个统一模型 UniIF,用于所有分子的逆折叠。**研究人员在蛋白质设计、RNA 设计和材料设计等多个任务上进行了全面实验,以证明 UniIF 的有效性。结果表明,UniIF 在所有任务上都达到了最先进的性能。
相关研究以「UniIF: Unified Molecule Inverse Folding」为题,入选顶会 NeurIPS 2024。
研究亮点:
* 研究提出的统一模型 UniIF 为一般分子逆折叠提供了一种多功能且有效的解决方案
* 该模型从两个层面进行统一:在数据层面提出了所有分子的统一块图数据形式,包括局部坐标系的构建和几何特征的初始化;在模型层面引入几何块注意力网络,捕捉所有分子的三维相互作用
* 研究人员证明了所提出的方法在蛋白质设计、RNA 设计和材料设计三大任务上都优于最先进的方法,这一成就可能对机器学习、药物发现和材料科学界产生积极影响

**数据集:选择对应数据集进行三种任务实验
蛋白质设计任务中**,研究人员在 CATH4.3 数据集上评估 UniIF。该数据集按 CATH 拓扑分类代码分割,产生 16,631 个训练样本、1,516 个验证样本和 1,864 个测试样本。
为了评估泛化能力,研究人员采用时间划分策略,考虑到一些基线使用预训练的 ESM2 模型,存在数据泄漏的风险。时间划分评估将特定日期之前的数据分配给训练集,而将该日期之后的数据分配给测试集。对于结构的时间划分评估,使用 CASP15 数据集,其中包含在训练期间未见过的新晶体结构;对于序列的时间划分评估,使用 NovelPro 数据集,该数据集包含 2023 年 11 月 23 日之前 30 天内发布的 76 个蛋白质序列,结构由 AlphaFold 2 预测。
**RNA 设计任务中,**研究人员在 RDesign 收集的数据集上进行 RNA 实验,该数据集包含 2,218 个 RNA 三级结构,这些结构根据其结构相似性分为训练集 (1,774 个结构)、测试集 (223 个结构) 和验证集 (221 个结构)。由于数据样本数量较少,研究人员报告了 3 次独立运行的中位数恢复率及其标准差。
材料设计任务中,研究人员在 CHILI-3K 数据集上评估 UniIF,该数据集由单金属氧化物衍生的纳米材料图构成。数据集包括 53 种金属元素和一种非金属元素 (氧),共计 3,180 个图,6,959,085 个节点和 49,624,440 条边。
模型架构:用于一般分子逆折叠的统一模型 UniIF
如下图所示,研究人员提出了一个用于一般分子逆折叠的统一模型。
① 该模型将所有类型的分子 (All Molecules) 转换为块图——对于大分子 (Macromolecules),使用基于氨基酸和核苷酸的预定义框架;对于小分子 (Small molecules),通过一层 GNN 学习每个块的局部框架;
② 使用几何特征提取器 (Geometric Featurizer) 初始化几何节点特征 (Node feature) 和边特征 (Edge features);
③ 提出了块图注意力层 (Block Graph Attention),基于此构建块图神经网络 (Block Graph Neural Network) 以学习表达丰富的块表示;
④ 最后展示 UniIF 在多种任务 (Tasks) 上均能取得具有竞争力的结果,包括蛋白质设计、RNA 设计和材料设计。
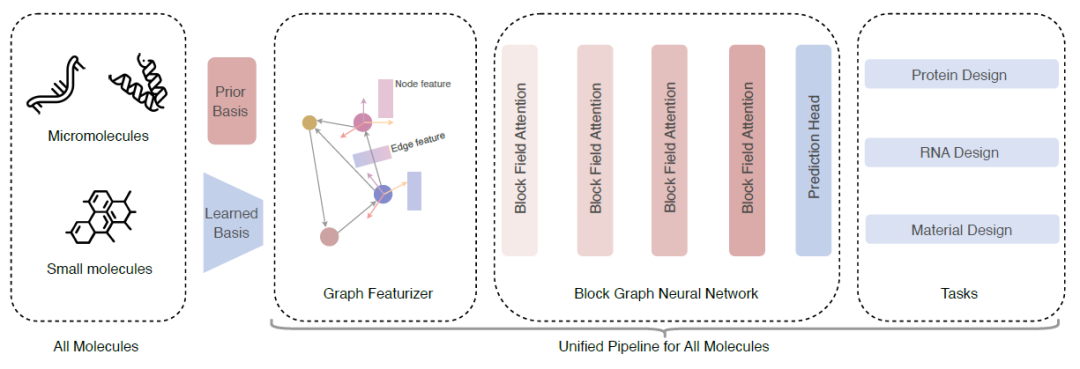
图:UniIF 模型整体框架
**构建块图:**该模型架构的第一步是引入块图来表示所有类型的分子,关键在于将不规则的原子集合 (大小各异) 转换为规则的块表示 (固定大小)。研究人员引入基于框架的块表示,以统一对所有分子的建模,一个块包含等变框架和不变特征向量,局部框架包含轴矩阵和位移向量。对于大分子,轴矩阵是基于氨基酸和核苷酸预定义的;而对于小分子,因为小分子没有先验的共同结构模式,需要学习轴矩阵。给定一个包含 n 个块的分子,研究人员使用 kNN 算法构建块图。
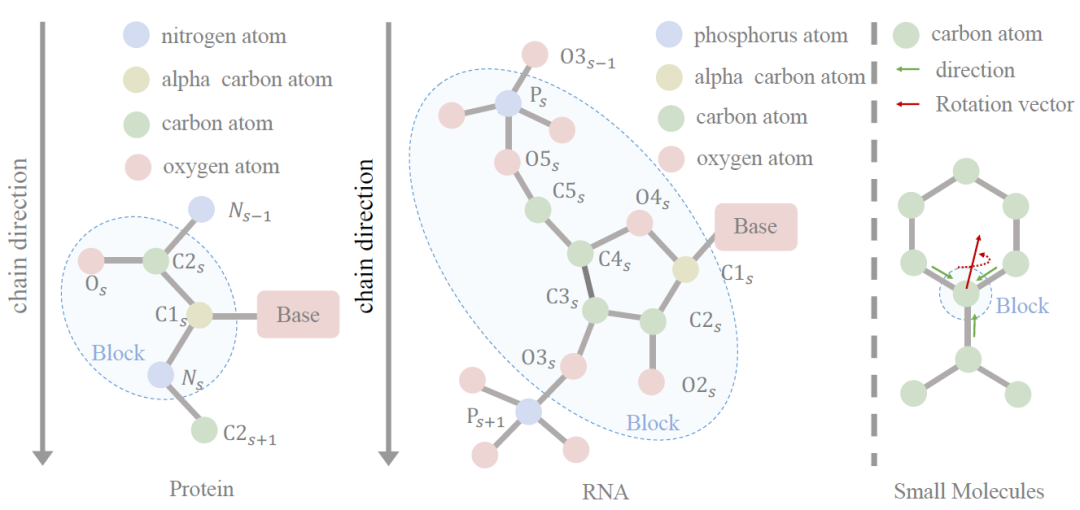
图:不同分子的结构单元,基本构建单元包括氨基酸、核苷酸和原子
**块图特征提取:**对于小分子,无法使用预定义的局部框架,因此研究人员需要为每个原子学习局部框架——即给定一个分子 ,其使用一层 GNN 来初始化原子表示,然后使用几何特征提取器初始化几何节点特征和边特征。
块图注意力模块:研究人员引入了几何块注意力网络,包括几何交互、交互注意力和虚拟长期依赖模块,以捕捉所有分子的三维交互。
研究结果:UniIF 在所有任务上都优于最先进的方法
研究人员通过多个逆折叠任务和消融研究展示了 UniIF 的有效性,包括:
* 蛋白质设计 (T1):设计能够折叠成目标结构的蛋白质序列
* RNA 设计 (T2):设计能够折叠成目标结构的 RNA 序列
* 材料设计 (T3):从已知材料结构中发现稳定的组成
① 蛋白质设计 (T1)
蛋白质设计旨在设计能够折叠成目标结构的蛋白质序列,研究人员在不同设置 (有和没有 ESM2) 和多个数据集 (CATH4.3、CASP、NovelPro) 下提供了结果。如下表所示:使用不包含 ESM2 的纯逆折叠模型,UniIF 在所有数据集上实现了最佳性能,证明了其有效性。
*LMDesign 和KWDesign 包含ESM2;StructGNN、GraphTrans、GCA、GVP、AlphaDesign、ProteinMPNN 和 PiFold 不包含 ESM2
在 CATH4.3 上,由于基线模型较强,整体提升有限,但时间划分评估突显了 UniIF 在泛化能力上的优势,UniIF 以更少的可学习参数超越了强基线 PiFold。在时间划分评估中,UniIF 以显著的优势超过了所有基线,包括基于 ESM2 的方法。在包含新序列的 NovelPro 上,UniIF 的表现优于使用 ESM2 进行序列优化的 LMDesign 和 KWDesign——这表明 UniIF 具有优越的泛化能力,对于实际应用至关重要。
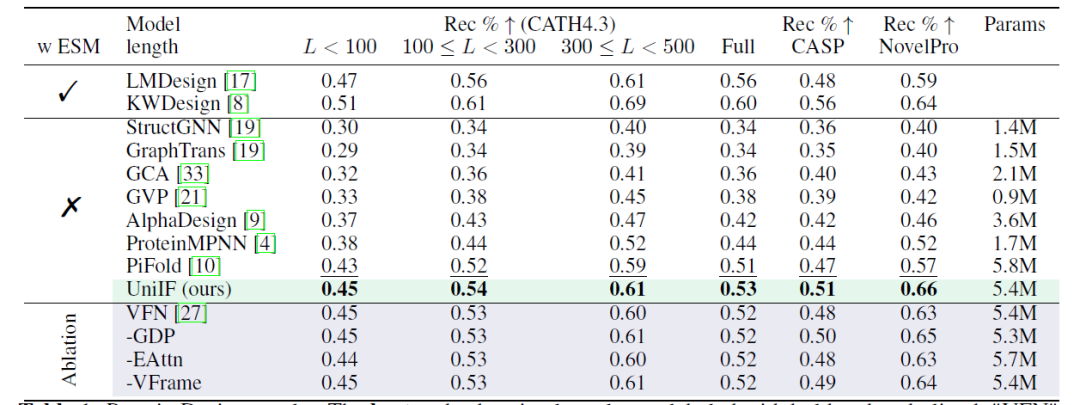
蛋白质设计结果 (最佳和次优结果用加粗和下划线标注)
② RNA 设计 (T2)
RNA 设计的目标是设计能折叠成目标结构的 RNA 序列。如下表所示,UniIF 在所有情况下均取得最佳性能,这一提升是显著的,因为之前强大的基线模型如 PiFold 仅在蛋白质设计上表现突出。据悉,UniIF 是第一个在蛋白质和 RNA 设计任务中都实现最先进性能的模型,证明了其多功能性和有效性。
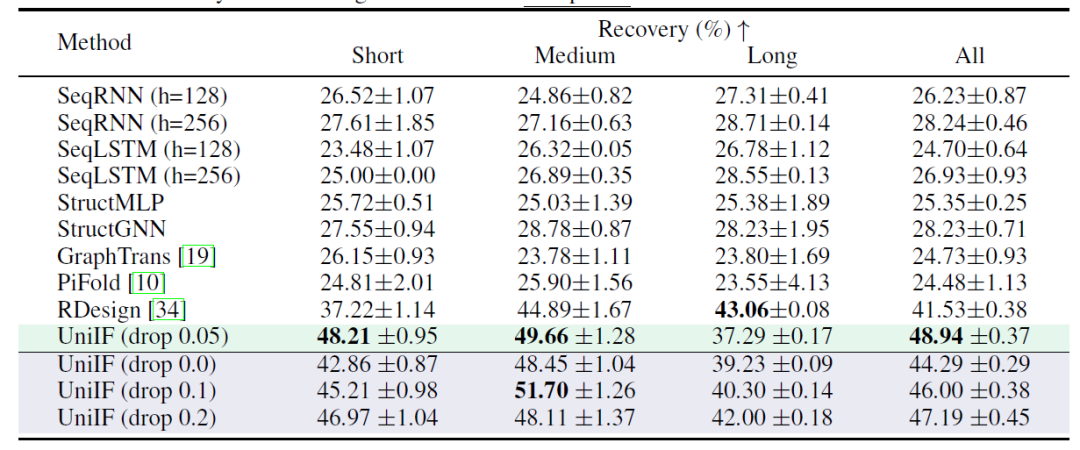
RNA设计的恢复结果 (最佳和次优结果用加粗和下划线标注)
③ 材料设计 (T3)
从已知材料结构中发现稳定的原子组合对于新材料的发现至关重要,所以研究人员也评估了 UniIF 在这一新任务上的表现。如下表所示,UniIF 显著超越所有基线模型。
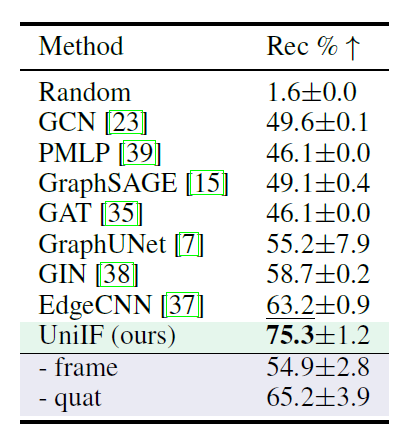
表:CHILI-3K 结果
④ 案例研究
在下图中,研究人员展示了设计的蛋白质和 RNA 序列。此外,其使用 AlphaFold 3 将设计的序列重新折叠成结构——真实结构(灰色)、PiFold 结构(绿色)和 UniIF 结构(粉色)进行了对齐和比较。研究人员观察到,UniIF 在恢复率和均方根偏差 (RMSD) 方面都取得了改善,证明了其在逆折叠任务中的有效性。

图:设计示例,将真实结构 (灰色)、PiFold 结构 (绿色) 和 UniIF 结构 (粉色) 对齐
UniIF 模型对 AlphaFold 3 形成进一步补充
通用分子学习在近年来受到越来越多的关注,RoseTTAFold All-Atom (RFAA) 和 AlphaFold 3 是两个在该方向取得显著成功的代表性模型。
2024 年 3 月 7 日,David Baker 在 Science 发布了题为「Generalized biomolecular modeling and design with RoseTTAFold All-Atom」的研究论文。该团队开发了 RoseTTAFold All-Atom (RFAA),它可以将氨基酸和 DNA 碱基基于残基的表示与所有其他基团的原子表示相结合,从而对包含蛋白质、核酸、小分子、金属和给定序列和化学结构的共价修饰组件进行建模。
2024 年 5 月 9 日,Demis Hassabis、John Jumpe 等人在 Nature 发表了题为「Accurate structure prediction of biomolecular interactions with AlphaFold 3」的研究论文。该研究推出了 AlphaFold 3,这一最新模型能预测含有蛋白质数据库 (Protein Data Bank) 内几乎所有分子类型的复合物的结构,包括配体 (小分子)、蛋白质、核酸 (DNA 和 RNA) 如何聚集在一起并相互作用,以及预测翻译后修饰和离子对这些分子系统的结构影响,从而帮助科研人员在原子水平上精确地观察生物分子系统的结构。
细究这两种模型,RFAA 使用原子-键图表示小分子,而使用框架图表示大分子;AlphaFold 3 则采用双层表示,即原子表示和标记表示,适用于所有分子。标记概念就相当于前文所述的块概念,表示一组原子,如氨基酸或核苷酸。
GET 和 EPT 是最近提出的两个模型,采用块表示法同时适用于小分子和大分子,并引入了新的等变变换器骨架。与指定小分子原子-键图的 RFAA 不同,本文介绍的 UniIF 模型为所有分子类型采用统一块图,不需要原子-键图,而且该模型还为每个块引入了向量基,这一点与 AlphaFold 3 、GET 和 EPT 都不同。
由于在一定程度上解决了构建通用分子模型的挑战,**UniIF 模型可以视作在 RoseTTAFold All-Atom 和 AlphaFold 3 等「前辈们 」分子结构预测方向取得进展的进一步补充。**未来,不断迭代的生物大模型将帮助研究人员重新认识生物世界、重新思考药物发现,从而造福于全人类。
来源: HyperAI超神经
