
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
近日,美国加州理工学院Sarah E. Reisman课题组报道了一种镍催化酰亚胺(imide)亲电试剂和(杂)芳基卤化物的不对称还原交叉偶联反应,从简单的起始原料合成了一系列对映富集的α-芳基戊二酰亚胺(α-arylglutarimides),具有良好的收率和对映选择性。文章链接DOI:10.1021/jacs.4c09327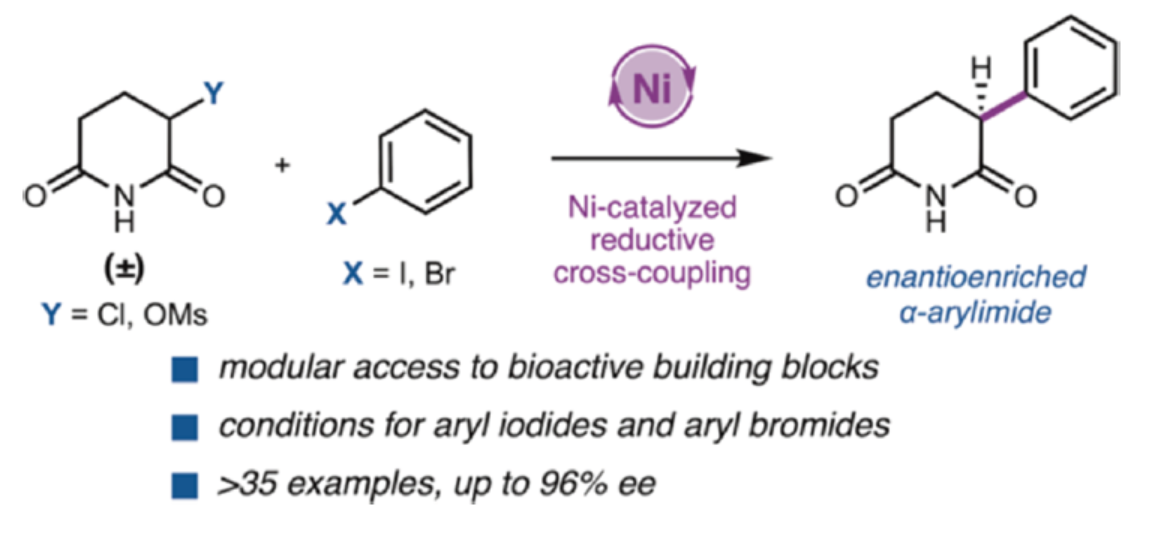
(图片来源:J. Am. Chem. Soc.)
正文
α-芳基酰亚胺骨架广泛存在于各种生物活性化合物中(Figure 1a)。特别是,α-芳基戊二酰亚胺在靶向蛋白质降解领域引起了广泛关注。α-芳基戊二酰亚胺是沙利度胺、来那度胺和其他α-N-杂环戊二酰亚胺等分子设计的类似物,它们与E3连接酶Cereblon结合,诱导靶蛋白的泛素化。前期,α-芳基戊二酰亚胺的合成路线通常需要多步反应。其中,利用α-芳基氰基酯进行酸性水解和环化构建α-芳基戊二酰亚胺是一种常见的方法(Figure 1b)。同时,2,6-二苄氧基吡啶通过交叉偶联进行官能团化,然后同时进行去苄基和氢化反应,也是合成α-芳基戊二酰亚胺是另一种常见的方法(Figure 1c)。然而,上述的两种方法均需使用预官能团化的底物且操作繁琐。另外,上述两种方法均未能直接获得对映富集的α-芳基戊二酰亚胺。为了解决这些问题,近日,美国加州理工学院Sarah E. Reisman课题组报道了一种镍催化酰亚胺亲电试剂和(杂)芳基卤化物的不对称还原交叉偶联(RCC)反应,合成了一系列对映富集的α-芳基戊二酰亚胺,亲电试剂对的合理选择使得反应具有良好的收率和对映选择性(Figure 1d)。欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
(图片来源:J. Am. Chem. Soc.)
首先,作者以3-氯哌啶-2,6-二酮1与4-碘甲苯2a作为模型底物,进行了相关反应条件的筛选(Table 1)。当以NiBr2·diglyme(10 mol %)作为镍预催化剂,(R)-L1(11 mol %)作为配体,Zn0(1.5 equiv)作为终端还原剂,TMSCl(0.8 equiv)作为添加剂,在DMA/THF的混合溶剂中18 oC反应2 h,可以85%的收率得到α-芳基戊二酰亚胺产物5a,ee为94%。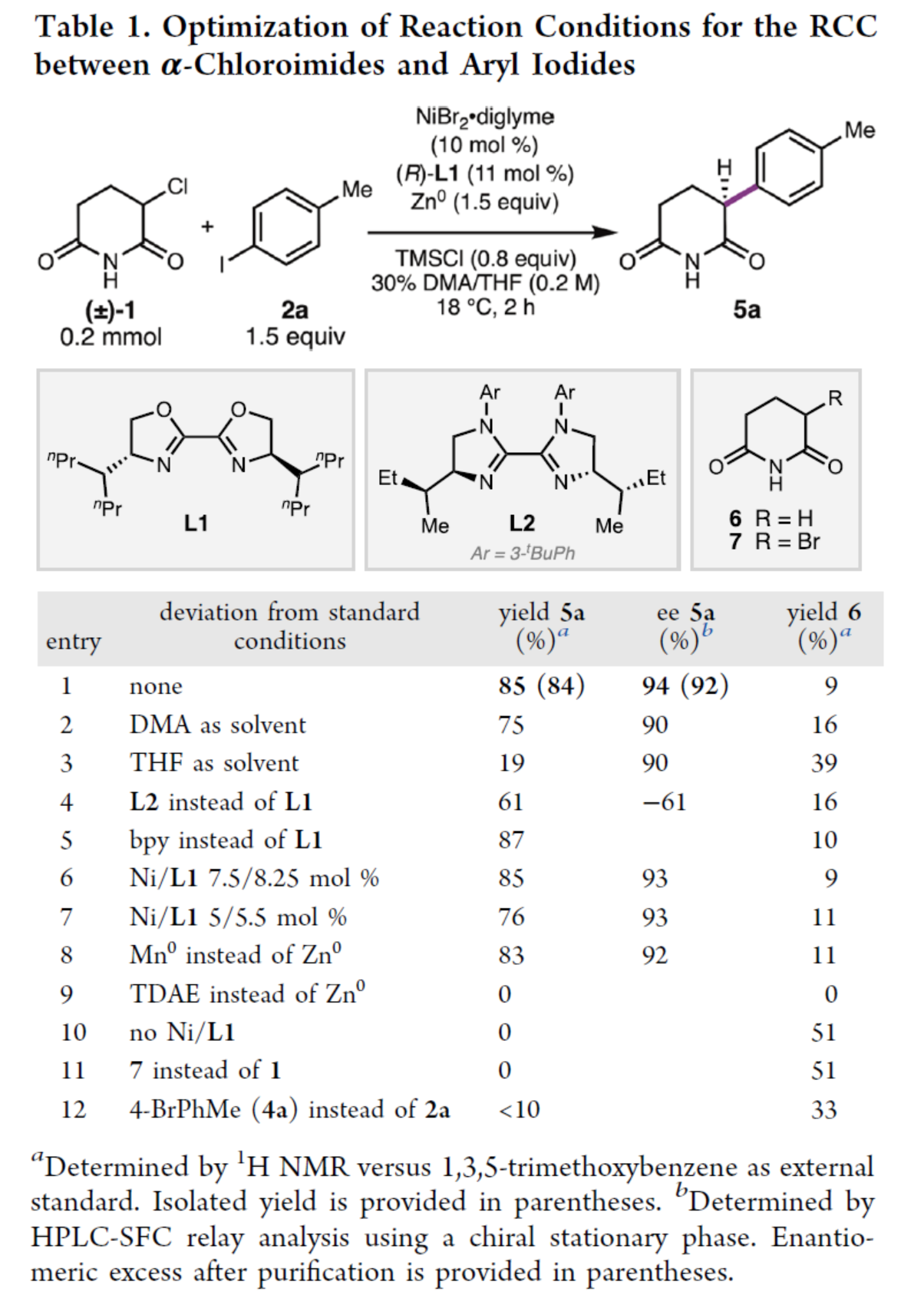
(图片来源:J. Am. Chem. Soc.)
在获得上述最佳反应条件后,作者对底物范围进行了扩展(Figure 2)。在Method A条件下,一系列不同电性取代的芳基碘,均可顺利进行反应,获得相应的产物5a-5v,收率为16-84%,ee为85-96%。其中,由于空间位阻的原因,导致反应的效率偏低,如5n、5o和5t。同时,碘代吡啶衍生物,也与体系兼容,获得相应的产物5x-5z和5aa-5ab,收率为9-56%,ee为90-93%。此外,由于缺电子的芳基溴化物应比相应的芳基碘化物经历更慢的氧化加成反应,作者认为,芳基溴化物可能在反应中表现更好。通过下述优化的条件(见Table 2,即Method B),一系列不同取代的芳基溴与杂芳基溴化物,也能够顺利进行反应,获得相应的产物5a-5i、5r、5s、5w、5x和5aa-5af,收率为36-72%,ee为63-90%。其中,杂芳基溴化物进行反应时,反应的对映选择性通常较低,这可能是由于这些更缺电子的产物在反应条件下外消旋化的倾向增加。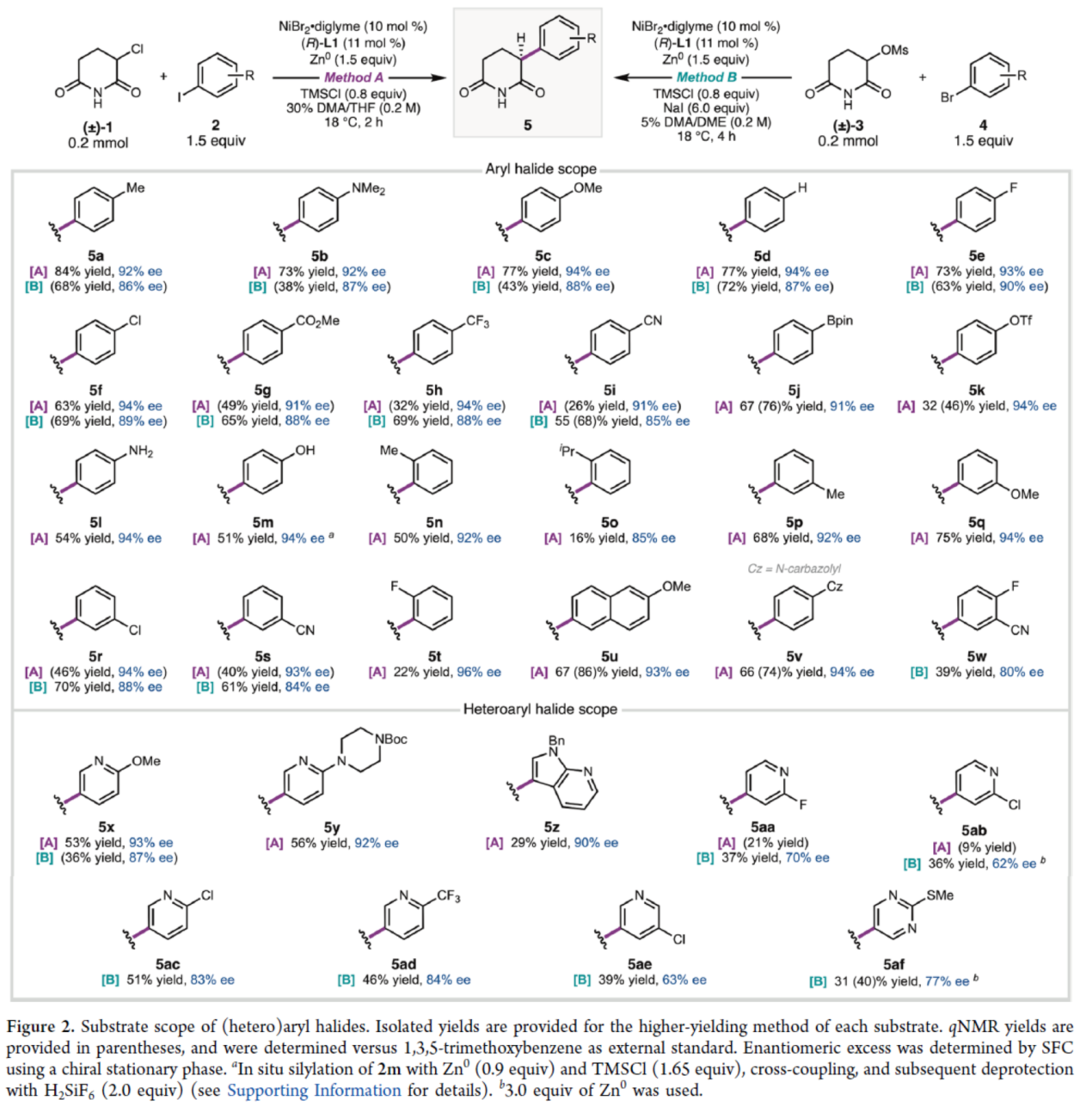
(图片来源:J. Am. Chem. Soc.)
带有α-磺酸酯的酰亚胺可以原位转为为α-卤代酰亚胺,这将有助于保持α-卤代酰亚胺的浓度较低,从而有效降低总活化速率。为此,作者以α-甲磺酸酯3与芳基溴化物4h作为模型底物,进行了相关反应条件的筛选(Table 2)。当以NiBr2·diglyme(10 mol %)作为镍预催化剂,(R)-L1(11 mol %)作为配体,Zn0(1.5 equiv)作为终端还原剂,TMSCl(0.8 equiv)作为添加剂,NaI(8.0 equiv)作为添加剂(诱导RCC反应),在DMA/THF的混合溶剂中18 oC反应4 h,可以74%的收率得到α-芳基戊二酰亚胺产物5h,ee为90%。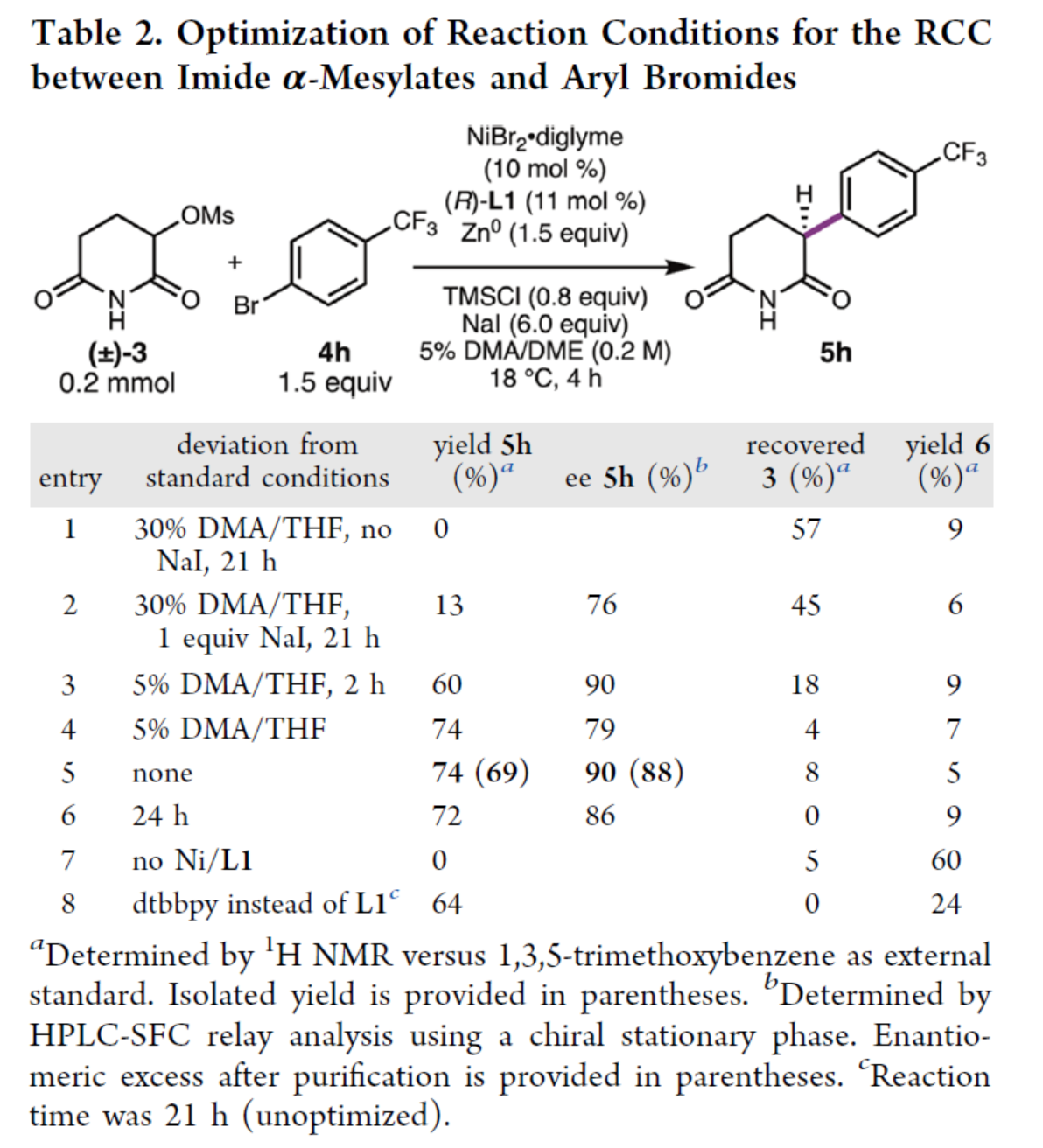
(图片来源:J. Am. Chem. Soc.)
同时,作者发现,几种α-取代酰亚胺也可作为有效的偶联底物(Figure 3a)。7元环状α-氯酰亚胺8a,可以62%的收率与87% ee得到产物9a,而相应的5元环状底物仅以36%的收率与60%ee得到产物9b。β-取代酰亚胺8c和N-对甲氧基苄基酰亚胺8d,也能够顺利进行反应,获得相应的产物9c(收率为65%,ee为86%)和9d(收率为50%,ee为92%)。其次,在α′-位含有额外手性中心的α-氯酰亚胺,当分别使用(R)-L1和(S)-L1时,可分别以良好的非对映体选择性获得相应的反式和顺式偶联产物9e(Figure 3b)。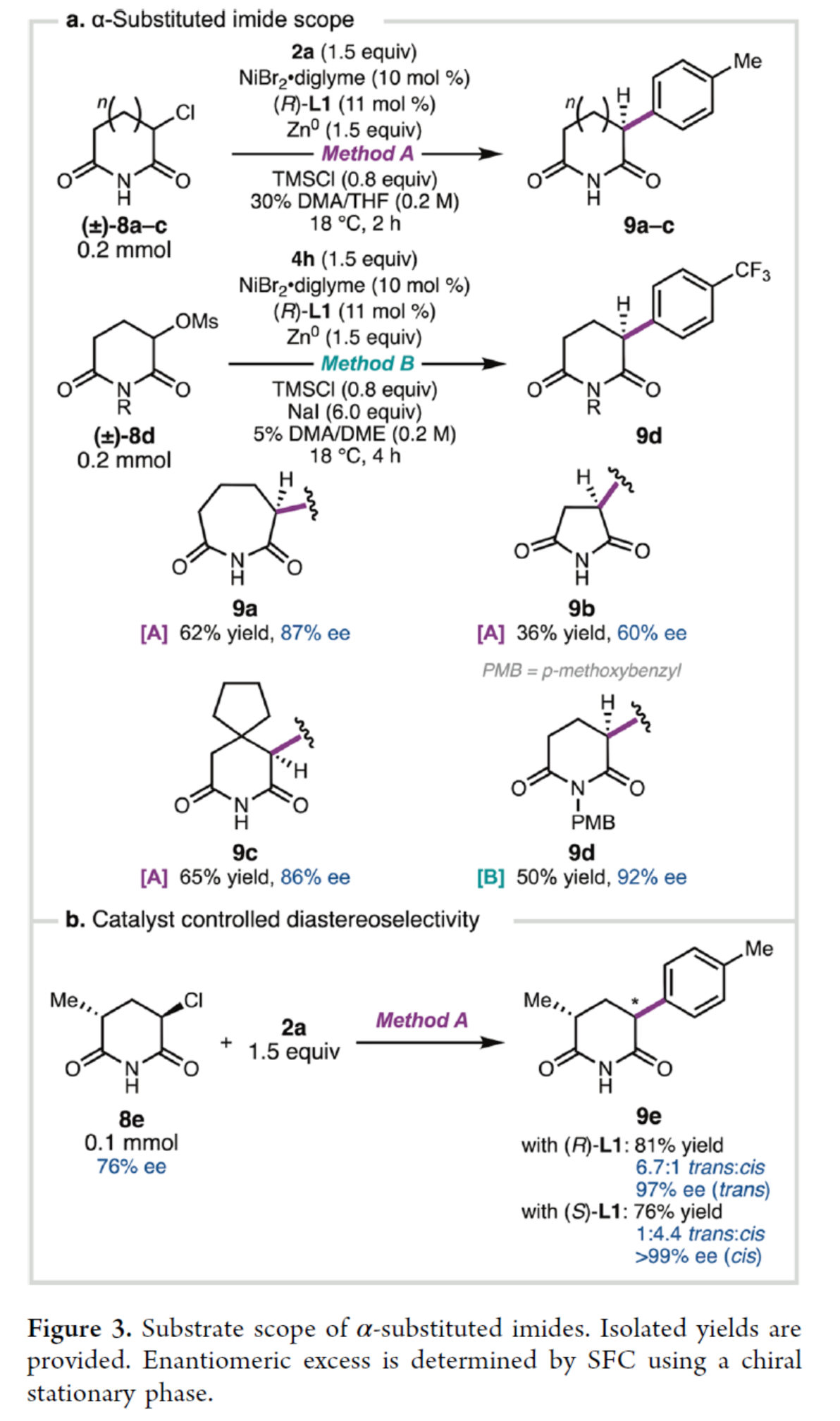
(图片来源:J. Am. Chem. Soc.)
为了进一步了解反应的机理,作者对反应的过程进行了监控(Figure 4a)。在前期,芳基碘2h不断积累(可能是镍催化卤化物交换的结果),但在后期逐渐衰减。相比之下,在反应过程中没有观察到由3衍生的α-卤代酰亚胺,这表明这些物种如果形成,会被迅速消耗。其次,3在NaI/DMA/DME条件下反应,可以43%的收率得到α-碘代酰亚胺11(Figure 4b)。作者认为,3在原位转化为α-卤代酰亚胺,其通过L1·Ni配合物进行卤素原子转移(XAT)或被Zn0还原以生成自由基。
基于前期相关文献的总结,作者提出了一种可能的反应机理。首先,在L1·NiBr2预催化剂还原后,L1·NiIX可与芳基卤化物进行氧化加成以及还原,生成L1·NiIIArX配合物。同时,α-卤代酰亚胺可以通过L1·NiIX配合物的XAT活化,也可以通过Zn0还原,生成α-imidoyl自由基。这种自由基可以被L1·NilArX配合物捕获,并通过还原消除,从而得到最终产物。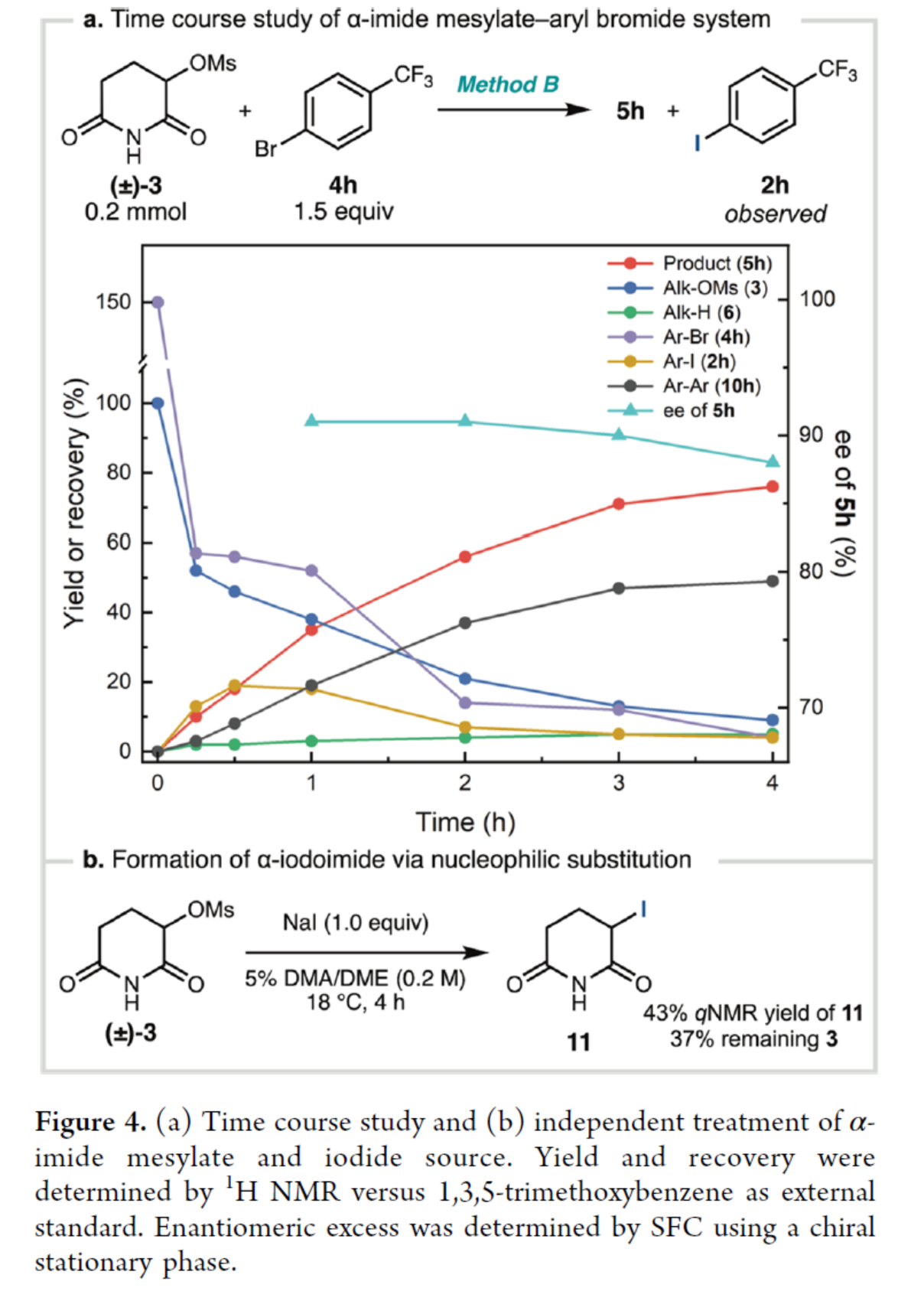
(图片来源:J. Am. Chem. Soc.)
紧接着,作者还使用成对电解对电化学驱动的交叉偶联反应进行了初步研究(Scheme 1)。研究结果表明,RCC在一个无隔膜电解槽中可以32%的收率进行,表明了镍催化剂能够活化α-氯酰亚胺1。然而,该反应的转化率和Faraday效率较低。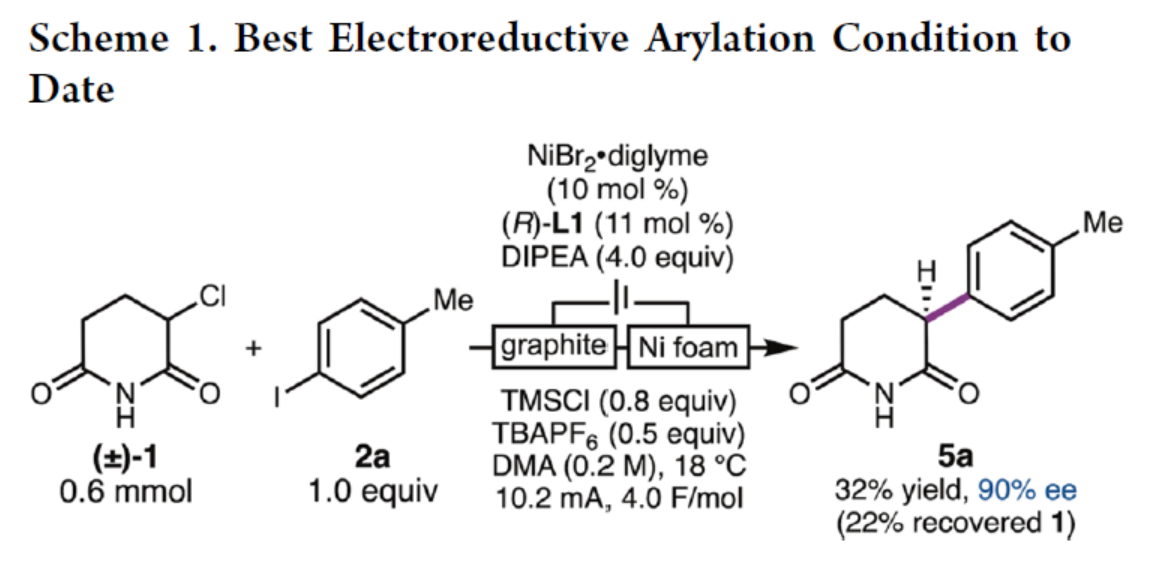
(图片来源:J. Am. Chem. Soc.)
最后,作者将产物5置于pH 7缓冲水溶液中,对产物5的对映选择性随时间的变化进行了研究(Figure 5)。研究表明,在对位具有吸电子基的α-芳基酰亚胺在24小时内迅速发生外消旋,如5h和5i。相反,对位带有供电子基的产物是稳定的,24小时后观察到的外消旋化最小。在邻位具有取代基的产物(5n和5t)比在对位具有取代基的产物(5a和5e)表现出更优的构型稳定性,这可能是由于去质子化位点的空间位阻增加导致。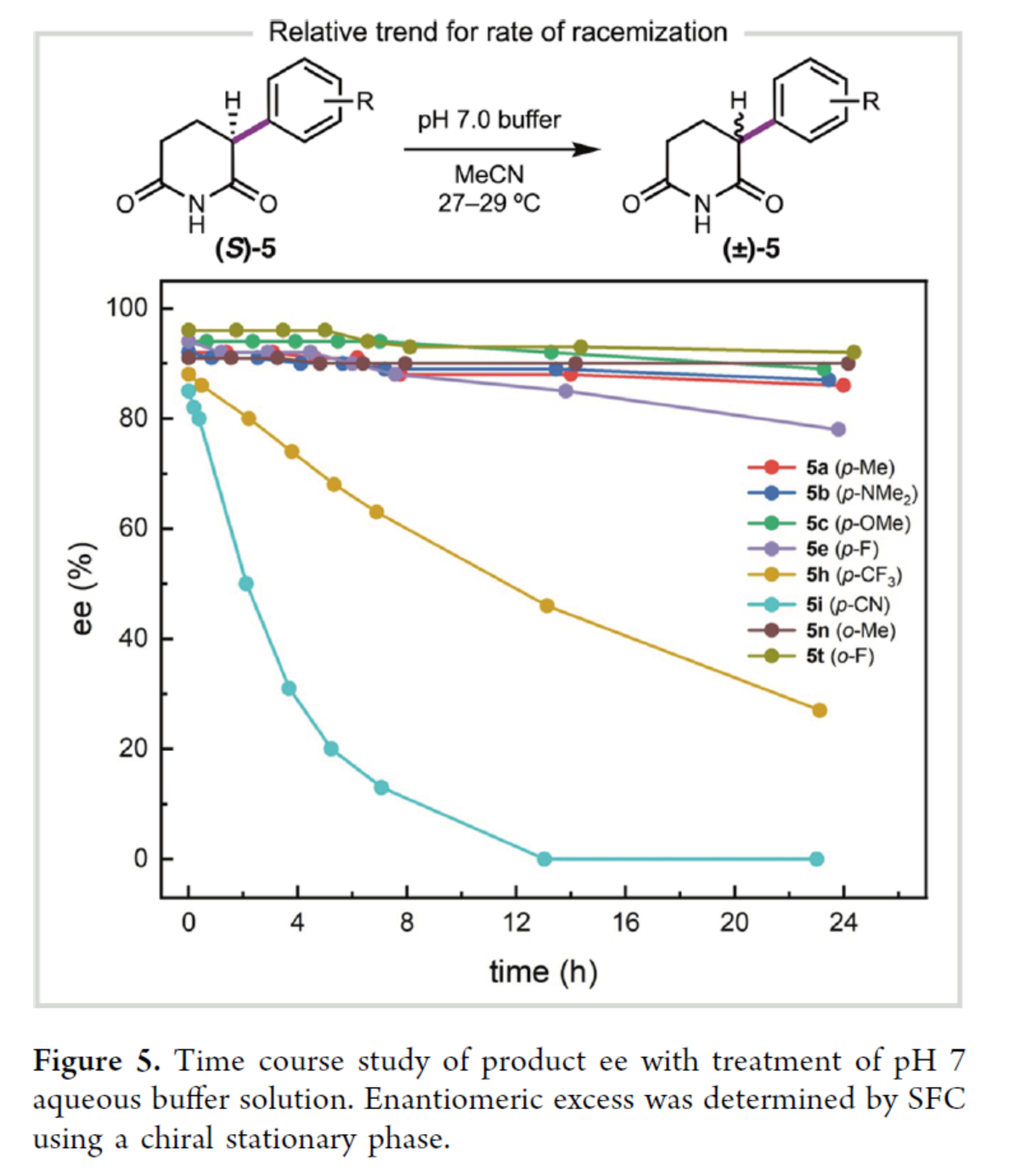
(图片来源:J. Am. Chem. Soc.)
总结
加州理工学院Sarah E. Reisman课题组报道了一种镍催化α-取代酰亚胺和(杂)芳基卤化物的不对称还原交叉偶联反应。对于含有供电子取代基的芳烃,芳基碘和α-氯酰亚胺的RCC均具有优异的性能。对于具有吸电子取代基的芳烃,芳基溴和α-酰亚胺甲磺酸之间的RCC表现更为有效。该策略可以有效地偶联广泛的(杂)芳基卤化物。这种转化使得高度对映富集的α-芳基戊二酰亚胺的合成变得容易,这可以作为制备PROTACs和其他生物活性分子的有力工具。
文献详情:
Ni-Catalyzed Asymmetric Reductive Arylation of α‑Substituted Imides.
Li-Ming Chen, Chungkeun Shin, Travis J. DeLano, Alba Carretero-Cerdan, Golsa Gheibi, Sarah E. Reisman*.
J. Am. Chem. Soc. 2024
https://doi.org/10.1021/jacs.4c09327
来源: 化学加
