
 科普中国公众号
科普中国公众号
 科普中国微博
科普中国微博

 帮助
帮助
 化学加
化学加 
导读
近日,韩国科学技术院(KAIST) Yoonsu Park教授报道了一种光催化策略,将呋喃的氧原子与氮基团交换,在单个分子间反应中直接将呋喃转化为吡咯类似物。该反应具有高兼容性,从高分子复杂性的天然呋喃后期功能化可得到原本难以获得的吡咯。机理表明,通过单电子转移的极性反转可在室温下启动氧化还原中性原子交换过程。文章DOI:10.1126/science.adq6245
正文
呋喃和吡咯是代表性的五元π电子等排体,它们的区别分别在于杂原子氧和氮。传统方法通常会制备呋喃基和吡咯基化合物用于SAR研究,但依赖于每种化合物的线性、平行合成,不可避免地增加了所需的合成工作量。评估杂原子效应的直接方案在很大程度上难以实现,在这种情况下,图1A中描绘的理想化呋喃单原子交换将能直接获得高级合成中间体或天然产物的各种杂芳族类似物。将氧原子形式上转化为其他原子可在不改变环大小的情况下提供结构同类物,并且这种杂环到杂环的转化将快速构建功能分子库,从而能够对以前无法获得的化合物进行SAR分析。欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
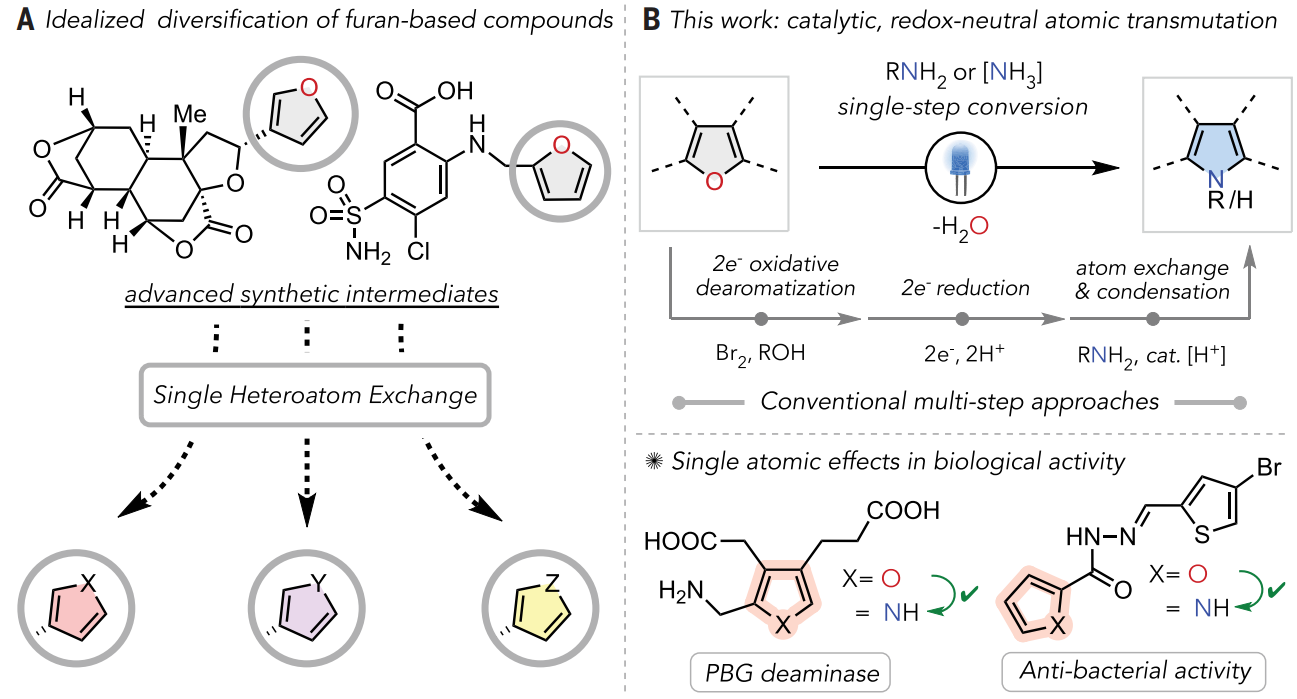
图1. 五元芳香杂环中的单原子交换(图片来源:Science)
这种“逆合成简单”方法的一个关键挑战在于芳香杂环的固有稳定性。所提出的原子交换可以分为三个典型过程:芳香性的初始破坏、杂原子的取代和芳香性的重建。Yoonsu Park教授课题组报道了一种光催化转化方法,该方法可在单一反应中将呋喃直接转化为吡咯,如图1B所示。该反应已成功应用于广泛的呋喃和氮源,光子能量消除了对化学计量的高能试剂的需求,水是唯一的副产物。
如图2A所示,在氨流下用活性氧化铝在450 °C下热解呋喃时,首次观察到呋喃直接转化为吡咯。使用多孔沸石材料进一步提高了反应性,其中高温下呋喃的Lewis酸活化被认为是驱动不利偶联的关键。
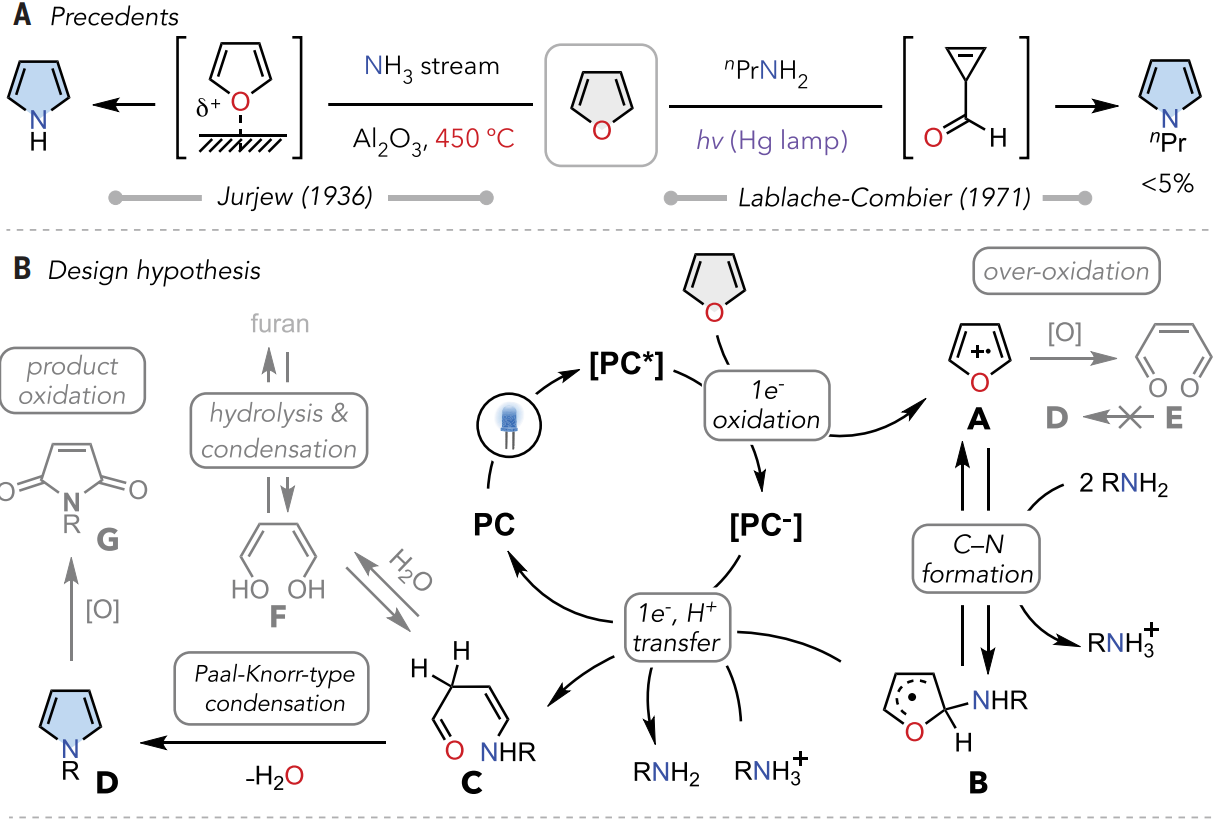
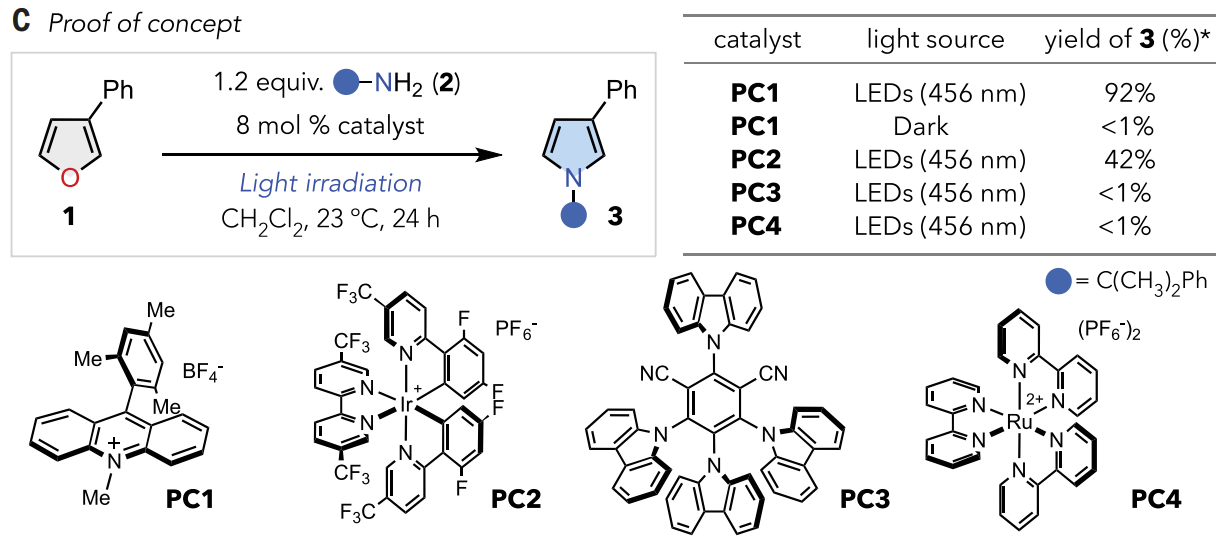
图2. 设计原理和概念验证研究(图片来源:Science)
图2B中所示的本文的关键假设是通过光氧化还原催化下的极性反转策略诱导分子间亲核试剂-亲核试剂偶联。具体而言,该反应由光激发下氧化催化剂激发态[PC*]对芳香呋喃进行单电子氧化引发。氧化的呋喃阳离子A变得缺电子,反转的极性促进胺与A的亲核加成,顺利形成加合物B。随后从还原的[PC-]进行电子转移和质子转移,得到单重态开环中间体C,其可以通过Paal-Knorr型缩合形成吡咯。
根据机理假设,使用3-苯基呋喃1作为模型底物,以枯基胺2作为氮源考察了催化反应性(图2C)。经过广泛的筛选,发现在蓝光照射下,基于吖啶的光催化剂PC1在室温下以92%的产率将1转化为其吡咯基类似物3。未观察到图 2B中所示的过度氧化或副产物,这表明该反应具有高度选择性。添加 B(C6F5)3时反应产率提高。二氯甲烷为最有效的溶剂。
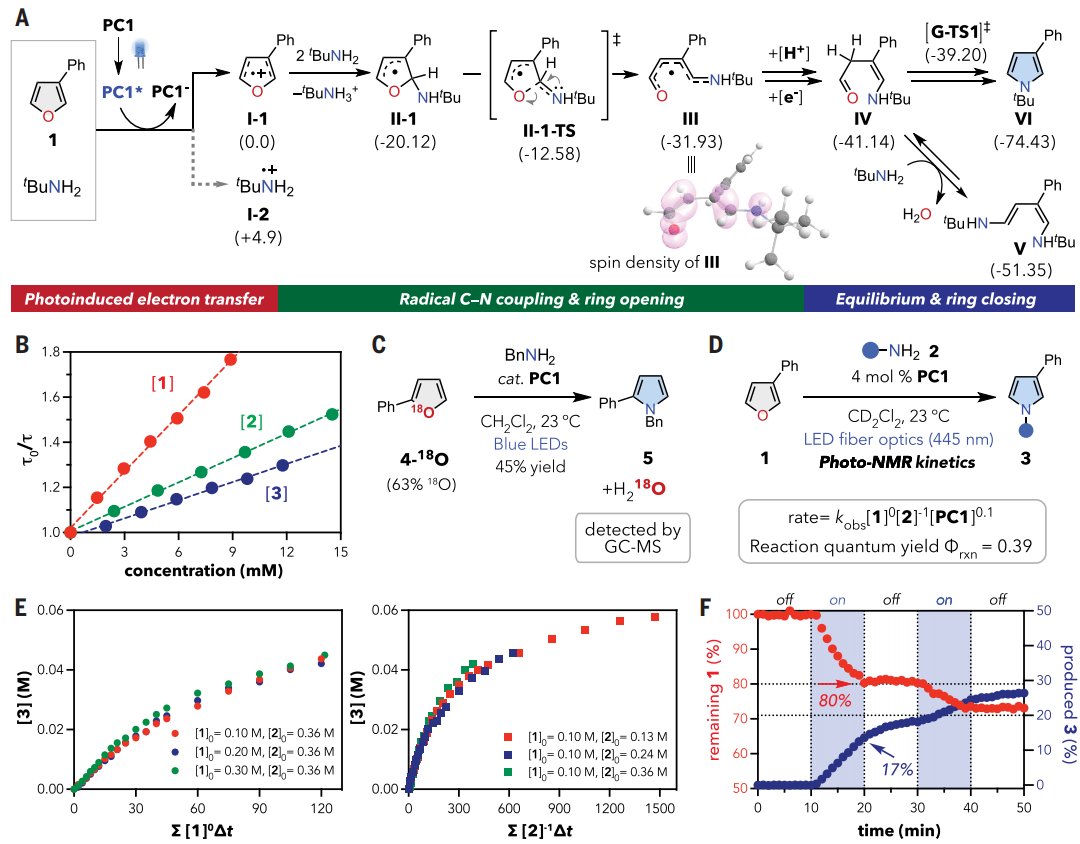
图3. 机理研究(图片来源:Science)
作者还进行了一系列理论和实验研究,以了解原子团交换过程的异常反应性。采用DFT计算来考察图2B中提出的途径的能量学。反应能量曲线总结在图3A中,初始步骤涉及PC1的光激发,这提供了高度氧化的激发态 PC1*。激发态可以通过呋喃或胺还原淬灭,同时形成还原的PC1-。自由能计算表明,呋喃(I-1)的单电子氧化在热力学上比叔丁胺(I-2)的单电子氧化所需能量低4.9 kcal/mol。氧化时极性反转为1,产生的呋喃自由基I-1可能与叔丁胺的C2或C5位结合。考虑了加合物II-1和II-2的区域异构体,计算预测II-1的稳定性更高,主要是由于苄基稳定效应。
中间体II-1随后通过II-1-TS发生C-O键均裂得到III,活化能垒为7.5 kcal/mol。稳定的III可以在PC1-单电子还原后提供醛IV,然后进行质子转移和酮-烯醇互变异构化。随后IV的分子内缩合通过Paal-Knorr吡咯合成产生吡咯,并释放化学计量的水为单一副产物。根据上述计算结果,作者通过激发态的实验淬灭研究了光引发过程。如图3B所示,表明呋喃1是PC1*最有效的淬灭剂。循环伏安实验进一步证实了呋喃1在热力学上比烷基胺更有利于氧化,而3的不可逆氧化发生在比1更低的起始电位下。
作者通过一系列实验研究了整体催化机理。如图3C所示,呋喃上的氧富含18O(4-18O),质谱分析在粗反应混合物中检测到富含18O的水,证实了转化的氧化还原中性和可持续性。呋喃1和光催化剂PC1 观察到了饱和动力学,而烷基胺2以负一级行为延缓了总反应速率。图3F中所示的开/关灯实验提供了有力的证据来合理解释速率定律。
呋喃消耗速率和吡咯形成速率之间的差异可以通过以下方式合理解释:(i)呋喃的光诱导消耗、(ii)开环中间体的积累和(iii)在没有光子能的情况下缓慢的Paal-Knorr型缩合。VTNA中2的负一级行为证实了这种机制场景-即开环中间体IV 可以隔离额外的烷基胺以形成非循环,如图3A中的二烯胺物种V,并且它可能以静止状态存在于溶液中。事实上,在辐射后在-30 °C下进行变温NMR监测显示推定物种的积累,在加热至室温后转化为吡咯产物3。DFT计算支持V在 Paal-Knorr机制相关中间体中的热力学稳定性。最后,测量到的反应量子产率为39%,表明光催化操作有效。天然丰度下的碳-13动力学同位素效应在吡咯产物的C2位上给出了1.016(1)的初级动力学同位素效应值。
接下来,作者评估了一般合成的适用性。如图4A所示,各种α-伯烷基胺,如苄胺衍生物(6-10),表现出优异的反应性。药学相关的哌啶部分适合优化条件(11),具有较长脂肪链的胺(12-15)顺利地以良好的产率经历了O-到-N的转化。重要的是,13的苯乙基部分在转化后可以很容易地脱除,得到游离的N-H吡咯14。酯(16)和缩醛(17)等多功能官能团完全兼容。此外,含有其他杂环的烷基胺,如噻吩、呋喃和游离吲哚,也适用于标准条件(18-20)。
本文的方案适用于一系列空间拥挤的烷基胺。如2-氨基茚满与呋喃1反应,以极好的产率生成吡咯21。对映体富集的苯乙胺以86%的产率生成相应的手性吡咯(R)-22,而不会破坏立体化学。合成完整性进一步通过23的立体特异性形成得到证实,该化合物未显示任何差向异构化的迹象。游离羟基(23、24)和张力氧杂环丁烷部分(25)与催化条件完全兼容。此外,各种α-叔烷基胺表现出优异的反应性。如图3A中用作 DFT计算模型的叔丁胺已成功应用(27),而体积庞大的1-金刚烷胺可获得类似的产率(28)。与2的10 mmol规模反应可得到2.43 g吡咯3,产率为93%,α-3°胺具有良好至优异的反应性(29-31)。富含π的炔基和空间位阻的哌啶基部分分别生成相应的吡咯32和33。
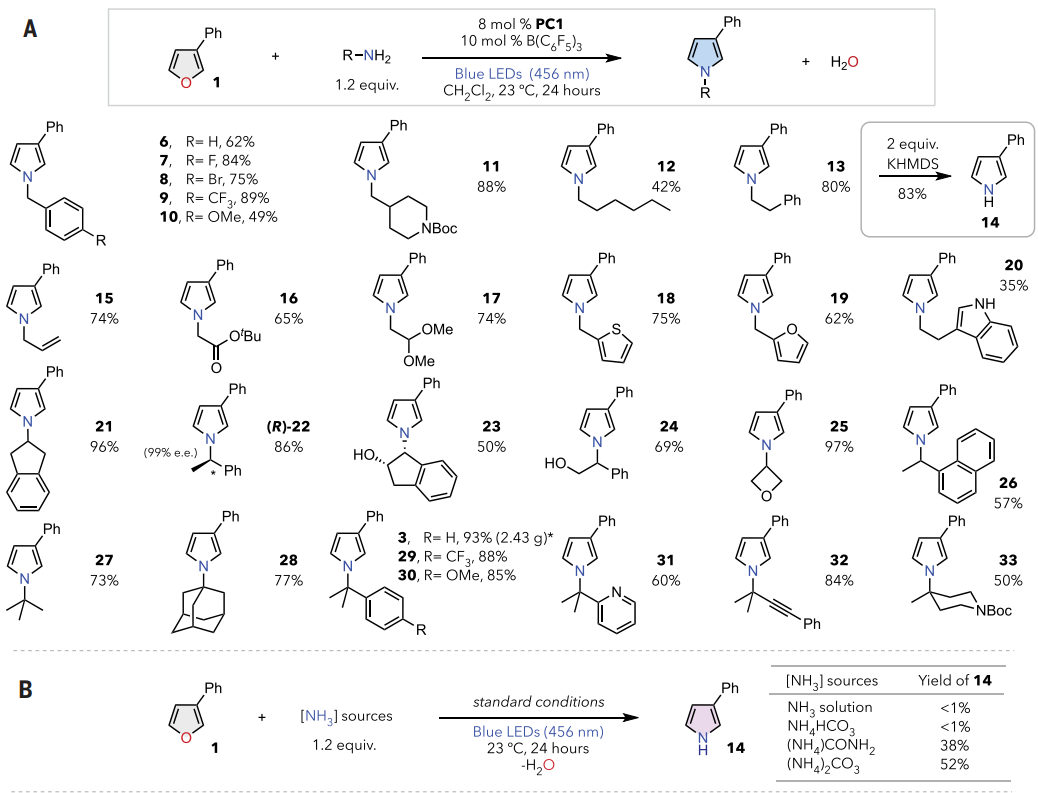
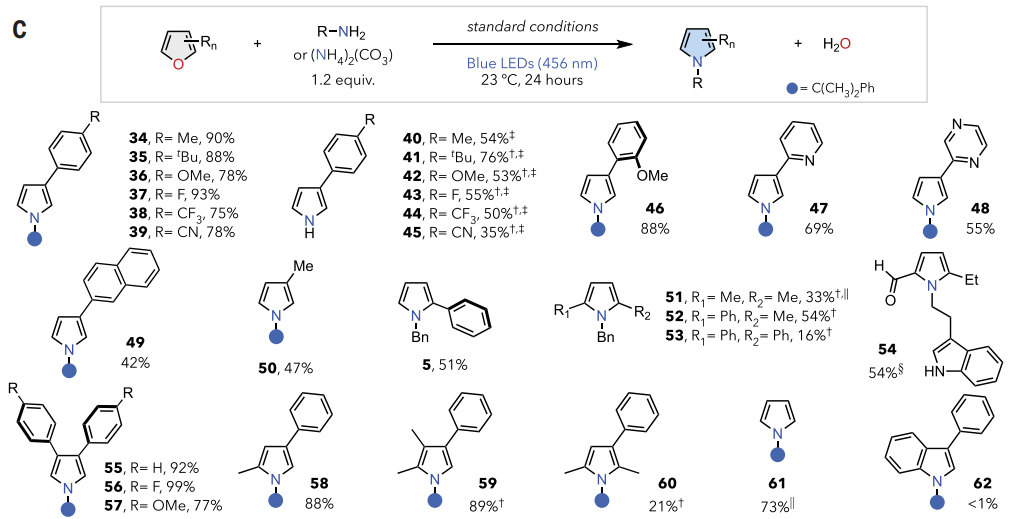
图4. 底物范围(图片来源:Science)
如图4B 所示,直接使用四氢呋喃中的氨溶液没有表现出明显的转化率,而含有氨基甲酸盐和碳酸根阴离子的铵盐分别以38%和52%的产率生成所需的吡咯14。
如图4C所示,电性变化通常会导致反应性略有不同(34-39,46)。以碳酸铵作为氮源可直接从相应的呋喃中得到各种 N-H 吡咯(40-45)。杂环取代基(包括吡啶基(47)和吡嗪(48)基团)不会干扰原子嬗变过程,3-萘基和-甲基取代基具有良好的耐受性(49, 50)。具有不同取代模式的呋喃也表现出良好的反应性。2-苯基呋喃与苄胺以良好的产率转化为相应的吡咯5。2,5-位的二取代呋喃产率不同,这可能是由于空间位阻所致(51-53)。5-乙基糠醛在色胺存在下进行单步交换(54),在3,4-和2,4-取代模式(55-58)下观察到良好至优异的反应性。虽然2,3,4-三取代呋喃具有优异的反应性(59),但在2,3,5-取代模式(60)下观察到较低的产率。石油原料、未取代的简单呋喃以73%的产率生成吡咯产物61。
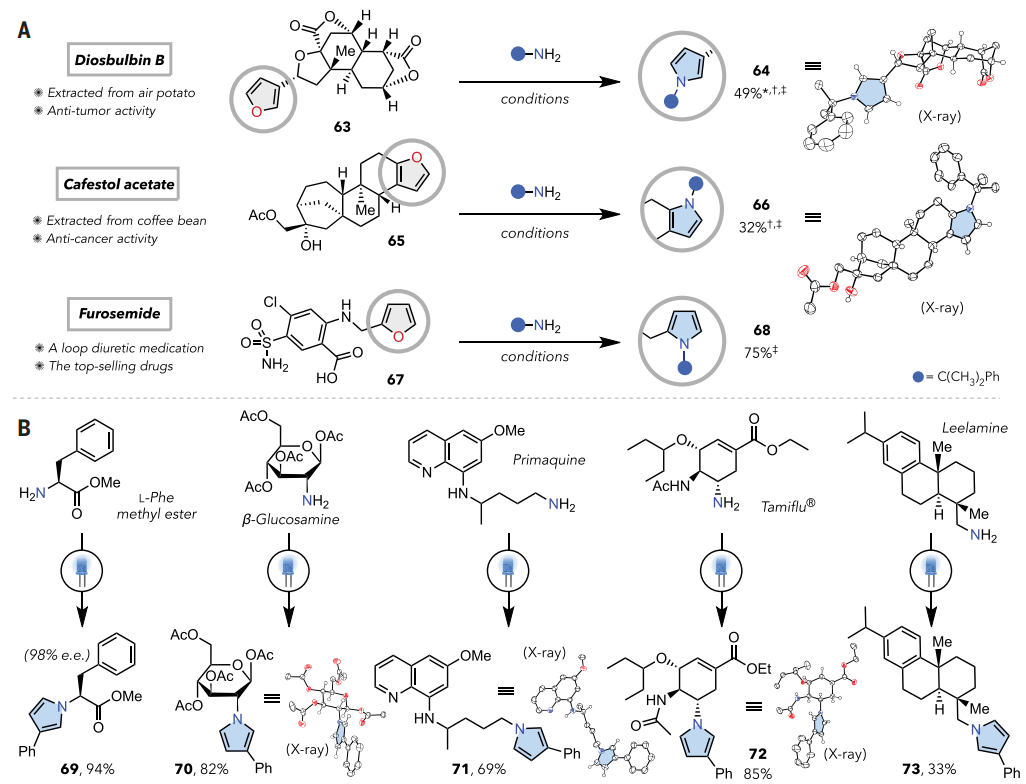
图5. 生物活性分子中的应用(图片来源:Science)
该方法的广泛兼容性促使作者测试了后期功能化中的应用,结果如图5所示。Diosbulbin B (63)是天然代谢物,具有抗肿瘤活性。应用本文的方案可以选择性转化为类似的吡咯化合物64,分离产率为49%。Cafestol乙酸酯65是一种从咖啡豆中提取的二萜类天然产物衍生物,当应用标准反应条件时,2,3-二取代呋喃部分成功转化为类似的吡咯66,产率为32%。呋塞米(67)(一种袢利尿药)也发现了类似的反应性。即使在羧酸、磺酰胺和氯基团存在的情况下,呋喃基部分也可以直接转化为其吡咯基类似物68,产率为75%。这些案例证明了O到N原子团交换在药物发现过程中的潜在效用,其中单步反应可以提供吡咯基类似物。
如图5B所示,手性苯丙氨酸甲酯在呋喃1存在下以94%的产率进行催化吡咯形成(69)。受保护的β-D-葡萄糖胺可得到相应的产物70,而其手性中心不会发生差向异构化。疟疾药物(伯氨喹)、达菲和天然产物(利拉明)也易反应(71-73)。
总结
Yoonsu Park教授报道了一种单步催化杂原子转化方案,该方案能够轻松地将呋喃化合物转化为相应的吡咯。本文的方案将广泛用于药物发现、材料科学等多个学科中。
文献详情:
Photocatalytic furan-to-pyrrole conversion.
Donghyeon Kim, Jaehyun You, Da Hye Lee, Hojin Hong, Dongwook Kim, Yoonsu Park*.
Science, 2024
https://www.science.org/doi/10.1126/science.adq6245
来源: 化学加
